![[Logo]](unilogo.gif)


![[Logo]](http://www.bioinf.uni-leipzig.de/pics/IfI.png)
- Bakterien auswählen und Genom herunterladen.
- Genom "bereinigen"
- paarweises/multiples Alignen
- 2 Methoden - 2 Ergebnisse
- RNAz und Ergebnisse
1. Bakterien auswählen und Genom herunterladen.
Wir haben uns bei der Auswahl der Bakterien für die Analyse von Kokken entscheiden. Nachdem wir uns kurz über diese Organismen informiert habe (siehe Einleitung) haben wir uns für folgende Bakteriengruppen entschieden:
|
|
Das Genom
Das Genom wurde unter Benutzung des NCBI Taxonomy Browser heruntergeladen und in den folgenden Dateien entsprechend der festgelegten Abkürzungen (siehe oben) gespeichert.Vorläufig wurde auch ein phylogenetischen Stammbaum erstellt anhand der Angaben des Taxonomy Browser erstellt.
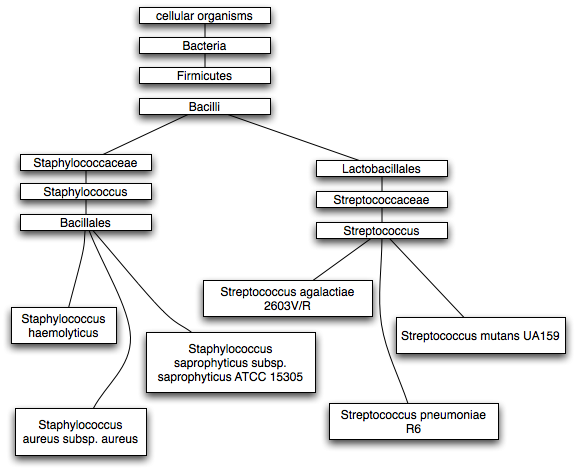
2. Genom "bereinigen"
Als nächstes wurden alle Protein-codierenden Regionen die in den Proteinannotationsdateien enthalten sind, im Genom mit dem Skript "cut_coding.pl" herausgeschnitten.Das Ergebniss sind Fasta Dateien die nun weiter berarbeitet werden können.
3. paarweises/multiples Alignen
Danach wurde mit Hilfe des Programms all_bz und dem phylogenetischen
Baum im Netwick Format (siehe
Material) ein bash Skript erstellt. Dieses Skript bildet die
paarweisen Alignments zwischen den Sequenzen mit Hilfe von Blastz. Das Skript
wurde mit folgendem Befehl erstellt:
‘all_bz - "((sp_ag sp_mu
sp_pn)(sy_au sy_ha sy_sa))" *.*.maf > &all_bz.log‘,
Da die Ausführung des Skriptes sehr lange dauert lief es über Nacht auf den Cluster PCs. Anschließend wurde das lokale multiple Alignment zwischen allen Alignments gebildet mittels des Programmes tba gebildet. Als Ergebniss erhielten wir diese tba.maf Datei.
4. 2 Methoden - 2 Ergebnisse
Ab diesem Schritt stehen 2 verschiedene Methoden zur Verfügung.Methode 1 besteht darin die erstellte tba.maf direkt an RNAz zu übergeben.
Methode 2 erstellt aus der tba.maf Datei für jedes Alignment eine einzelne ‘.aln‘ Datei.
Die Ergebnisse der beiden Methoden müsste theoretische gleich sein. Da wir uns nicht sicher sind welcher der beiden Algorithmen der bessere ist werden wir deren Ergebnisse am Ende noch überprüfen.
Bevor wir jedoch mit der weiteren Untersuchung beginnen konnten mussten wir die ‘maf‘ Datei noch ein wenig transformieren. Es wurde noch ein zusätzliches Perl Skript entwickelten, welches alle Alignments mit Score 0, sowie alle Alignments mit weniger als 20 Basenpaaren oder weniger als 3 Sequenzen entfernte.
4.1. Methode 1
Die erstellte tba.maf wird an rnazWindow.pl weitergeleitet. Dort wird sie auf RNAz vorbereitet und in Alignments mit max. 120 bp unterteilt. Die einzeln unterteilten Alignments überlappen sich jeweils um 40 bp. Dies geschieht durch ‘rnazWindow.pl‘.4.2. Methode 2
Nach dem aussortieren bestimmter Alignments aus der tba.maf werden alle Alignments getrennt in jeweils eine eigene Datei geschrieben.Aus den .aln Dateien wurden nun mittels eines Skriptes alle Gaps entfernt und mittels des Programms Clustal W die globalen Alignments berechnet. Wir erhielten die optimierten .aln Dateien und .dnd Dateien.
Mittels des Skriptes TrimRealignments.pl wurden nun Gaps am Anfang oder Ende eines Alignments entfernt.
Nun mussten wir ein weiteres Perl Skript entwickeln, welches die Alignment Dateien fü das Programm RNAz aufbereitet. Das Skript führte je nach Länge des Alignments entweder RNAz direkt, oder rnazWindow aus. Da RNAz nur Alignments mit weniger als 120 Basenpaaren verarbeiten kann. Die erstellten "Fenster" überlappen auch hier wie in Methode 1 um jeweils 40bp.
5. RNAz und Auswertung
RNAz berechnete nun die Bindungensenergie der Faltung der Sequenzen und ermöglichte dadurch eine genauere Analyse, ob eine jeweilige Sequenz tatsächlich einen wichtigen ncRNA Abschnitt enthielt.Die Abschnitte die durch RNAzWindow gesplittet wurden, mussten nun jedoch ersteinmal wieder zusammen gefürt werden. Dazu diente das Skript rnazCluster.pl. Dieses Programm erzeugte nun auch eine bildliche Darstellung des gefalteten Abschnitts und färbte die ncRNA Regionen.
Auswertung
Insgesamt haben wir 142 verschiedene Sequenz-Alignments an rnaZ übergeben.Desweiteren wurden nachdem rnaZ die Ergbenisse fertig berechnet hat noch eine spezielle Suche mit trna-Scan gestartet, sowie in den Datenbanken RFAM und NonCode bekannten Sequenzen gesucht.Auch die in den .frn Dateien von NCBI annotierten Bereiche wurden gesucht.Nachdem blasten wurde uns klar das die Methode 1 die besseren ist da sie mehr Ergebnisse und besser Annotationen liefert. Methode 2 wird daher in der Auswertung nicht weiter beachtet da ihre Ergebnisse nur eine kleine Teilmenge von Methode 1 sind.
Insgesamt wurden 141 Loci also 141 mögliche ncRNA's gefunden.
Von diesen Loci wurden 50 annotiert.
Übersicht welche Datenbank wieviel Annotationen lieferte:
| Datenbank | annotierte Loci | % |
|---|---|---|
| Rfam | 44 | 31 |
| NonCode | 5 | 03 |
| NCBI | 39 | 27 |
Die nicht annotierten Loci wurden mittels des Skriptes html_filter aus der results.html herausgeschnitten und die entsprechenden Sequenzen aus den Genomen extrahiert. Die Liste der nicht annotierten Loci im Fasta Format ist unter "Material" zu finden.
