Methoden
Ziel:
Anhand der snoStrip pipeline wurde innerhalb einer neuen D.plexippus Annotation die snoRNA Sequenzen bestimmt und nach eventuellen neuen snoRNA Gene gesucht. Anbei erfolgte eine Auswertung der gefunden snoRNA Gene.
Schritt 1: Finden der snoRNA-Gene aus der D.plexippus Annotation
Shell-Code:
grep 'snoRNA_gene' > snoRNA_gene.gff
grep 'snoRNA' > snoRNA.gff
- gff ist ein Dateiformat, welches Merkmale von Genen, Proteinen, DNA oder RNA Sequenzen beschreibt
- die zu beschreibene Merkmale folgen einer bestimmten Reihenfolge
- im weiteren werden wir die snoRNA-Gene benutzen, da es die Transkripte und genauere Informationen enthält
Schritt 2: Erstellen einer fasta-Datei anhand der gefundenen snoRNA Gene
aus dem älteren Genoms von Danaus plexippus.
Shell-Code:
bedtools getfasta -fi Danaus_plexippus_DanPle.1.0.fa -bed snoRNA_gene -fo dpl1.fa -s
- bedtools getfasta extrahiert Sequenzen aus einer Fasta-Datei für die definierten Intervalle aus einer gff-, bed- oder vcf-Datei
- wichtig ist, dass der Header in der Fasta-Datei der Chromosomspalte(bed) entspricht
- der FASTA-Header für die extrahierte Sequenz ist folgendermaßen formattiert: "chrom;start;end"
- "-s" wenn die Sequenz antisense Stränge beinhaltet, wird die reverse Sequenz komplementiert
- die Sequenzen wurden von der Datei "Danaus_plexippus_DanPle.1.0.fa" geladen; später als dpl1 bezeichnet
Start der snoStrip Annotations pipeline
Schritt 1 der snoStrip Annotations pipeline:
Schritt 3: Blasten der gefunden Sequenzen gegen die beiden Danaus plexippus Genome
Shell-Code (jeweils eine Zeile):
blastall -p blastn -d /scr/genomes/Metazoan-Animals/Danaus_plexippus/OTHER/daunaus_plexippus_supercontigs.fa -i dpl1.fa -m 8 -e 1e-10 > dpl3.blastout
blastall -p blastn -d /scr/genomes/Metazoan-Animals/Danaus_plexippus/ENSEMBL/Danaus_plexippus_DanPle.1.0.fa -i dpl1.fa -m 8 -e 1e-10 > dpl1.blastout
- BLAST erstellt ein lokales Alignment gegebener Sequenzen gegenüber einer bsp. Genomsequenz
- "-p" Programname; blastn für Nukleotide
- "-d" Datenbankenname
- "i-" zu blastende Datei
- "-m 8" Ausgangsdatei soll durch Tabulatoren getrennt sein
- E-Value ist 1e-10
- "danaus_plexippus_supercontigs.fa" wird von uns im nachfolgenden dpl3 genannt, auch die daraus abgeleitet Ergebnisse
Schritt 4: Umwandeln des Blastoutputs in bed-Dateien
Shell-Code (jeweils eine Zeile):
awk '{if($10>$9){print $2"\t"$9"\t"$10"\t"$1"\t"(($3*$4)/100)"\t-"}else{print $2"\t"$10"\t"$9"\t"$1"\t"(($3*$4)/100)"\t+"}}' dpl3.blastout > dpl3.bed
awk '{if($10>$9){print $2"\t"$9"\t"$10"\t"$1"\t"(($3*$4)/100)"\t-"}else{print $2"\t"$10"\t"$9"\t"$1"\t"(($3*$4)/100)"\t+"}}' dpl1.blastout > dpl1.bed
- die bed-Datei beinhaltet Informationen über Annotationen von Genen bzw. Sequenzen
- es beinhaltet drei benötigte und neun zusätzliche Spalten
- 1. Chromosomname, 2.Start, 3. Ende, 4.Name, 5. Score, 6. Strang
- es werden nur Gene mit einem Score von über 2000 benutzt
Schritt 5: Sortieren und Zusammenfügen der Datei dpl3.bed
Shell-Code:
sortBed -i dpl3.bed > dpl3_sort.bed
mergeBed -i dpl3_sort.bed > dpl3_sort_merge.bed
Schritt 6: Herausfinden der überlappenden snoRNA Gene mit Bedtools Intersect
Shell-Code:
bedtools intersect -a D.plexippus.gff -b dpl3_sort_merge.bed -wao
bedtools intersect -a dpl3_sort_merge.bed -b D.plexippus.gff -wao
- bedtool intersect sucht die Überlappungen zwischen zwei Sequenzen und gibt diese als bed-Datei aus
- "-a" vergleicht die Sequenzen zwischen Datei a mit Datei b
- "-b" vergleicht die Sequenzen zwischen Datei b mit Datei a
- "-woa" schreibt die Originale in die Ergebnisdatei und die Nummer der Basenpaare die überlappen
- die Datei "D.plexippus.gff" wurde uns von Stephanie Kehr zur Verfügung gestellt (aus den dpl1-Genom)
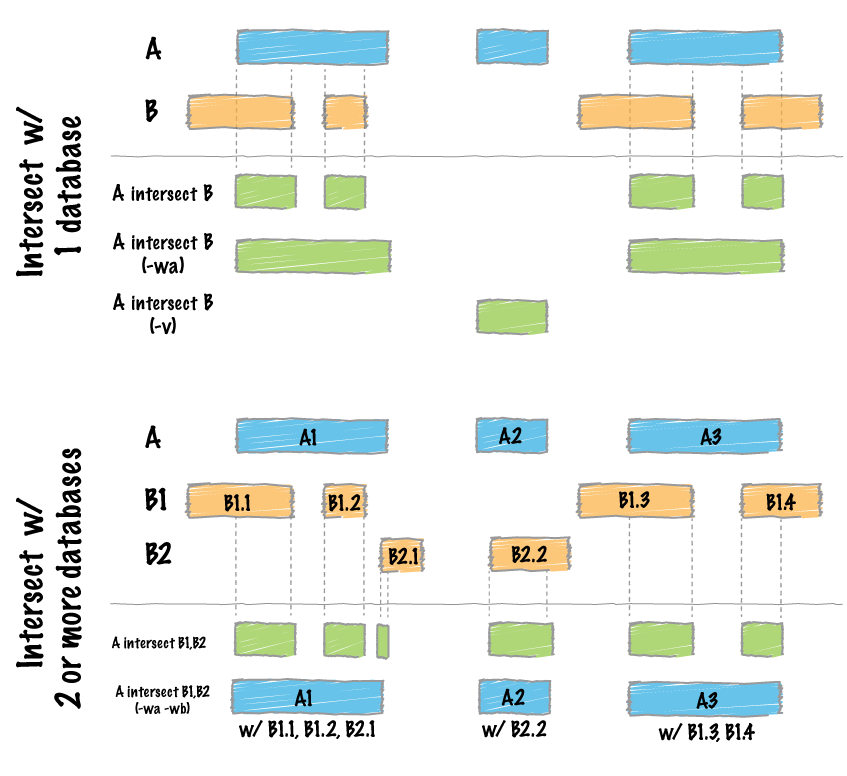
Bild 1: Darstellung der Vorgehensweise von bed intersect
Schritt 7: Anhand von Beispiel: Überlappen von Gen-ID in dpl1 und dpl3
- verwendete Gen-ID: JH387043
- Suchen der Gen-ID aus der dpl1.blastout in der dpl3.blastout Datei
- wenn Gen-ID in beiden Datein vorhanden, dann Auffinden der entsprechenden Sequenzen mit Hilfe von fastacmd
- dazu wurden die jeweiligen Genome benutzt
- für fastacmd wurden die jeweiligen DPSCF-ID's in dpl3 benutzt
- für fastacmd wurden die jeweiligen JH-ID in dpl1 benutzt
Shell-Code (jeweils eine Zeile):
fastacmd -d /scr/genomes/Metazoan-Animals/Danaus_plexippus/OTHER/daunaus_plexippus_supercontigs.fa -s 'DPSCF300789' -S '2' -L '5457,5539'
fastacmd -d /scr/genomes/Metazoan-Animals/Danaus_plexippus/OTHER/daunaus_plexippus_supercontigs.fa -s 'DPSCF300789' -S '2' -L '5357,5639'
fastacmd -d /scr/genomes/Metazoan-Animals/Danaus_plexippus/OTHER/daunaus_plexippus_supercontigs.fa -s 'DPSCF300470' -S '2' -L '49963,50045'
fastacmd -d /scr/genomes/Metazoan-Animals/Danaus_plexippus/OTHER/daunaus_plexippus_supercontigs.fa -s 'DPSCF300470' -S '2' -L '49863,50145'
fastacmd -d /scr/genomes/Metazoan-Animals/Danaus_plexippus/ ENSEMBL/Danaus_plexippus_DanPle.1.0.fa -i dpl1.fa -s 'JH387043' -S '2' -L '5457,5539'
fastacmd -d /scr/genomes/Metazoan-Animals/Danaus_plexippus/ ENSEMBL/Danaus_plexippus_DanPle.1.0.fa -i dpl1.fa -s 'JH387043' -S '2' -L '5357,5639'
- "-d" Datenbankeintrag
- "-s" Suchen des Eintrages (Gen)
- "-S" Richtung des Stranges: 1 - erster Eintrag (Nukleotidnummer) kleiner als zweiter; 2 - zweiter Eintrag ist kleiner als erster
- "-L" Angaben der Start und Stop Nukleotidnummern
- Schreiben der Sequenzen in eine Datei
Schritt 8: Sequenzalignment
Shell-Code:
muscle -in JH387043.fa -out JH387043.aln -clwstrict
Schritt 9: Umwandeln des clustal Alignment in Stockholm Datei
- wir benutzten ein Perl Script, von Stephanie Kehr um die clustalw-Datei in eine Stockholm Datei umzuwandeln
Shell-Code:
perl stockholm.pl JH387043.aln JH387043.aln.stk
 Praktikum Sequenzanalyse der Danaus plexippus snoRNA
Praktikum Sequenzanalyse der Danaus plexippus snoRNA