Protokoll zum Praktikum Bioinformatik WS 2011/2012
- Henrike Indrischek, Caroline Wilde -
TNF-alpha
Ziel des Praktikums war die Suche nach einem innerhalb der mRNA verschiedener Proteine konservierten Strukturmotiv, an welches die Proteinkinase PKR bindet und durch Autophosphorylierung aktiviert wird. Bei der PKR (Protein Kinase RNA-activated) handelt es sich um eine Serin/Threonin-Kinase, welche zwei dsRNA-Bindungsmotive und eine Kinase-Domaene besitzt. Strukturmotive, welche die PKR aktivieren, wurden unter anderem in den mRNA der Proteine Interferon-gamma (IFN), p23 und dem Tumor-Nekrose-Faktor-alpha (TNF-alpha) identifiziert. TNF-alpha ist auch unter den Namen Cachectin, tumor necrosis factor-alpha und dem Kuerzel TNFSF2 (tumor necrosis factor ligand superfamily member 2) bekannt. Osman et al. identifizierten 1999 ein cis-acting Element auf der 3'-UTR der TNF-alpha mRNA (bezeichnet als APRE, Aminopurin responsive element) als PKR-aktivierendes Strukturelement. Es war bereits bekannt, dass Aminopurin (AP) das Splicen von TNF-alpha beeinflusst. Mit Hilfe von genetischen Methoden und Restriktionsanalyse wurde eine phylogenetisch konservierte 17 bp umfassende Stem-Loop-Struktur als APRE-Region beschrieben, zu welchem PKR den zugehoerigen trans-acting Faktor darstellt. Desweiteren wurde durch Osman et al. nachgewiesen, dass die Aktivierung bzw. Inaktivierung der PKR vor allem von der Laenge und Position der Sequenz, weniger jedoch von der Sequenz der Stem-Loop-Struktur abhaengt. Das PKR-aktivierende Strukturelement der mRNA des IFN-gamma besitzt neben einer Stem-Loop-Struktur einen Pseudoknoten und einen Kink-Turn nachgewiesen und durch Cohen-Chalamish et al. gezeigt, dass dieser zur Aktivierung der PKR essentiell ist.
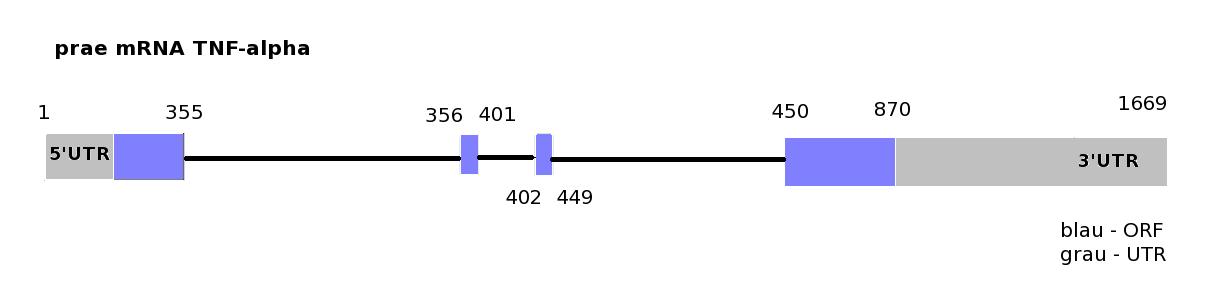
Die Struktur des TNF-alpha-Gens des Menschen (2761 bp) wurde mit Hilfe des UCSC Genome-Browsers und des NCBIs bei Suche unter dem Stichwort TNFSF2 ermittelt und ist folgend gezeigt. Das Gen besteht aus vier kodierenden Exons. Die nicht-translatierten Bereiche (UTR), welche jedoch transkribiert werden, befinden sich auf den Exons 1 und 4 (grau markiert). Die gesplicte mRNA mit polyA-Schwanz umfasst somit 1669 bp, die in das Protein mit 233 Aminosaeuren translatiert wird.
Methodik
Die nachfolgende Abbildung stellt die ausgeschnittene Sequenz von 870 bp dar (rote Linie).

Ergebnisse
Beim Alignen der Sequenzen verschiedener Organismen werden aufgrund der abweichenden Basenidentitaet ggf. gaps eingefuegt. Die Laenge der alignierten Sequenzen entspricht somit inklusive gaps nicht der eigentlich ausgeschnittenen Sequenzlaenge, dargestellt am Anfang und Ende des Alignments in folgender Abbildung. Das Sequenzalignment mit Clustalw zeigte einen sehr guten Basenkonsensus ueber weite Bereiche der UTR auf. Fuer die RNA-Strukturalignments wurden die Sequenzen deshalb so gekuerzt, dass sie mit einer einheitlichen Konsensussequenz anfingen und endeten, um die Strukturalignments einzelner Organismen vergleichen zu koennen (tuerkise Linie in obiger Abbildung).
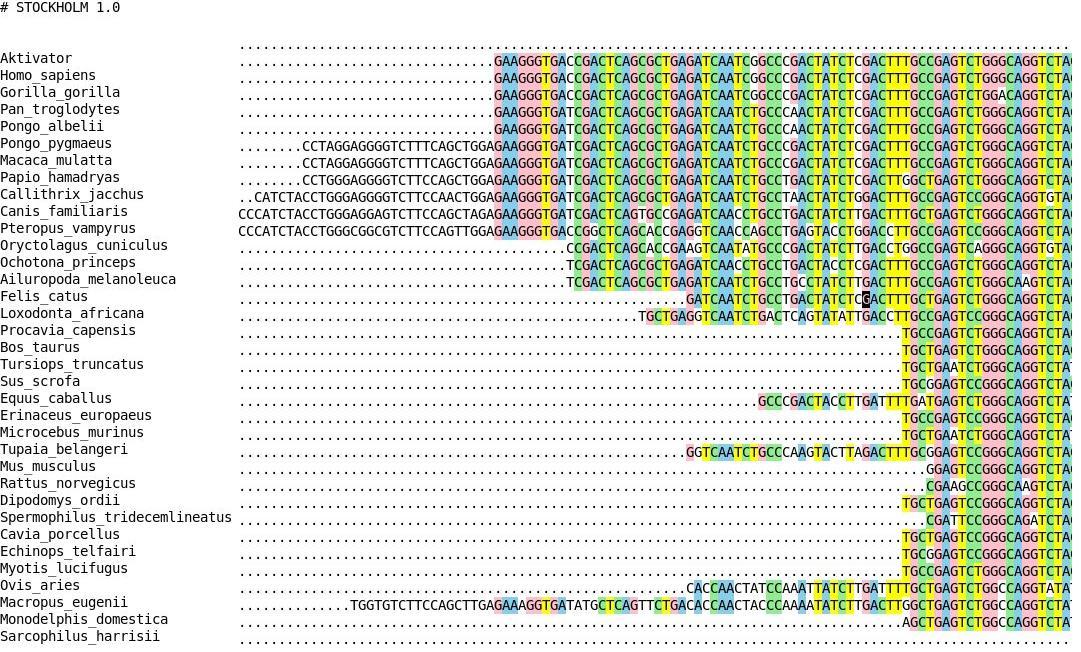
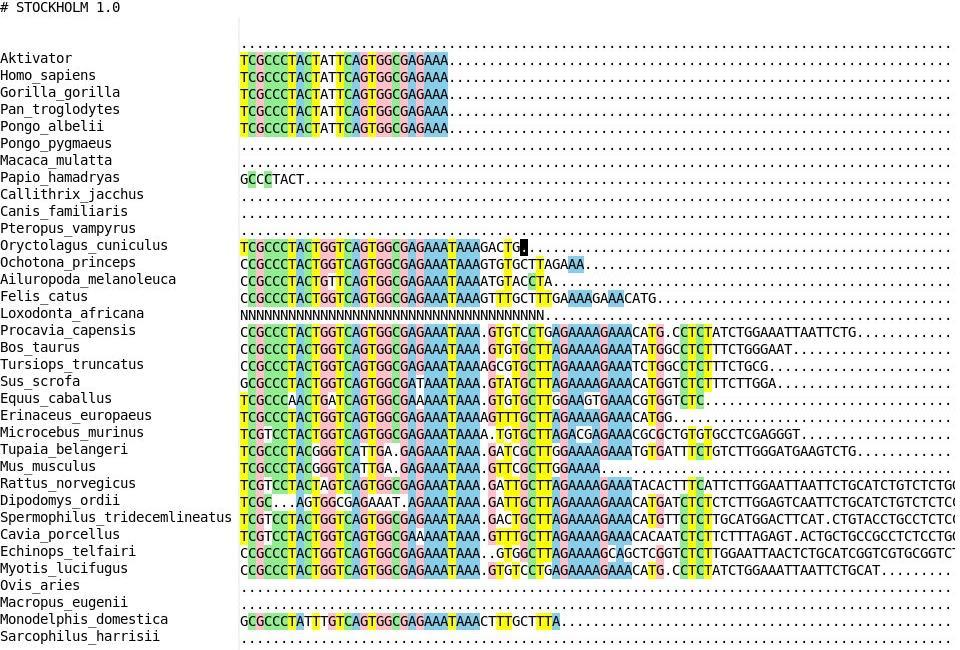

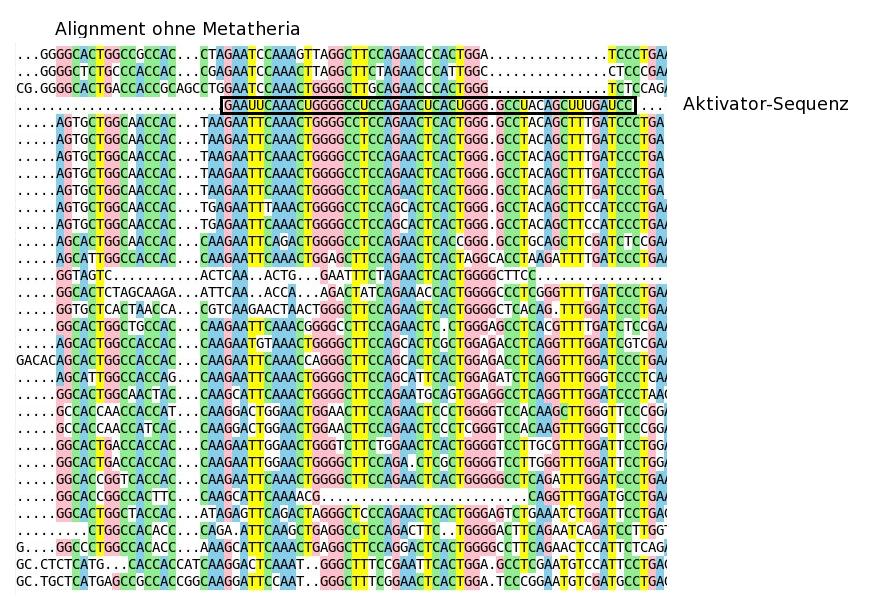
Mit Hilfe von Struktur- Alignments wurde die 3' UTR nach folgenden Aspekten untersucht: