 |
Bioinformatik-Praktikum
Modul Nukleinsäuren
|
 |
|


Durchführung
Step 0 - Vorarbeit
Zu Beginn galt es unseren Arbeitsplatz mit geeigneter Software auszustatten. Diese ist unter dem
Menüpunkt "Empfohlene Programme" auf unserer Linkseite zu finden.
Step 1 - Datenerhebung
Zu allererst stellten wir RNA-Sequenzen zusammen, welche wir im weiteren
hinsichtlich konservierter RNA-Sekundärsstrukturen untersuchen wollten.
Die Auswahl der Sequenzen erfolgte ausschließlich aus dem Datenbestand der
Universal Virus Database of the International Committee on Taxonomy of Viruses (ICTVdB) des
National Center for Biotechnology Information (NCBI).
Im Folgenden werden die Viren aufgeführt, aus welchen die verwendeten Sequenzen stammmen:
* Hierbei handelt es sich um Sequenzen die im Laufe der Untersuchungen
von weiteren Untersuchungen ausgenommen wurden,
da sie sich für die Untersuchungen als nicht geeignet erwiessen.
Aufgrund dessen werden nun im weiteren Verlauf mehrere Datenversionen angegeben,
einmal bezüglich 12 Sequenzen und einmal bezüglich 9 Sequenzen.
Dies geschieht bis zu dem Punkt in den Untersuchungen, wo festgestellt wurde,
dass die Verwendung der 9 Sequenzen günstigere Ergebnisse liefert.
Es wurden hier nur Beta-Retroviren ausgewählt.
Die ausgewählten Sequenzen liegen im FASTA-Format vor, welches auch das grundlegende Datenformat auf dem die Untersuchungen aufbauen, bildet. Allerdings musste vorher noch eine Headerkorrektur der von der ICTVdB geladenen Dateien erfolgen. Die aus der ICTVdB ermittelten Sequenzen wurden nun noch zur Weiterverarbeitung in einer einzigen Datei zusammengefasst.
| Accession-Number | Quellvirus | Sequenzlänge [bp] |
| AF033807 | Mouse mammary tumor virus | 8805 |
| AF033815* | Mason-Pfizer monkey virus | 8557 |
| AF105220 | Ovine pulmonary adenocarcinoma virus | 7455 |
| AF126467 | Simian retrovirus 2 | 8105 |
| AF228550 | Endogenous mouse mammary tumor virus Mtv1 | 9851 |
| AF228552* | Exogenous mouse mammary tumor virus | 9851 |
| M11841 | Simian SRV-1 type D retrovirus | 8173 |
| M12349 | Mason-Pfizer monkey virus | 8557 |
| M15122* | Mouse mammary tumor virus | 10125 |
| M16605 | Simian retrovirus 2 | 7759 |
| M23385 | Simian sarcoma virus | 8785 |
| M80216 | Ovine pulmonary adenocarcinoma virus | 7462 |
Es wurden hier nur Beta-Retroviren ausgewählt.
Die ausgewählten Sequenzen liegen im FASTA-Format vor, welches auch das grundlegende Datenformat auf dem die Untersuchungen aufbauen, bildet. Allerdings musste vorher noch eine Headerkorrektur der von der ICTVdB geladenen Dateien erfolgen. Die aus der ICTVdB ermittelten Sequenzen wurden nun noch zur Weiterverarbeitung in einer einzigen Datei zusammengefasst.
Step 2 - Sequenzalignment
Der erste Schritt bei der Analyse der ausgewählten Sequenzen ist das multiple Alignment aller Sequenzen.
Die Alignmentberechnung erfolgte zunächst mit dem Programm ClustalW.
Das Programm brachte folgende Alignment-Ergebnisse:
Das Programm brachte folgende Alignment-Ergebnisse:
- 12 Sequenzen Programmausgabe (ausgabe.txt, 3.6 KB)
- 9 Sequenzen Proggrammausgabe (ausgabe.txt,2.2 KB)
- 12 Sequenzen Alignment-Dateien(data.aln, 175 KB;
data.dnd, 227 byte)
- 9 Sequenzen Alignment-Dateien(data.aln, 135 KB;
data.dnd, 316 byte)
- Code2aln
- Programmausgabe (ausgabe.txt, 2 KB)
- Alignment-Dateien (data.aln, 119 KB)
- Dialign2
Step 3 - Phylogenetische Bäume
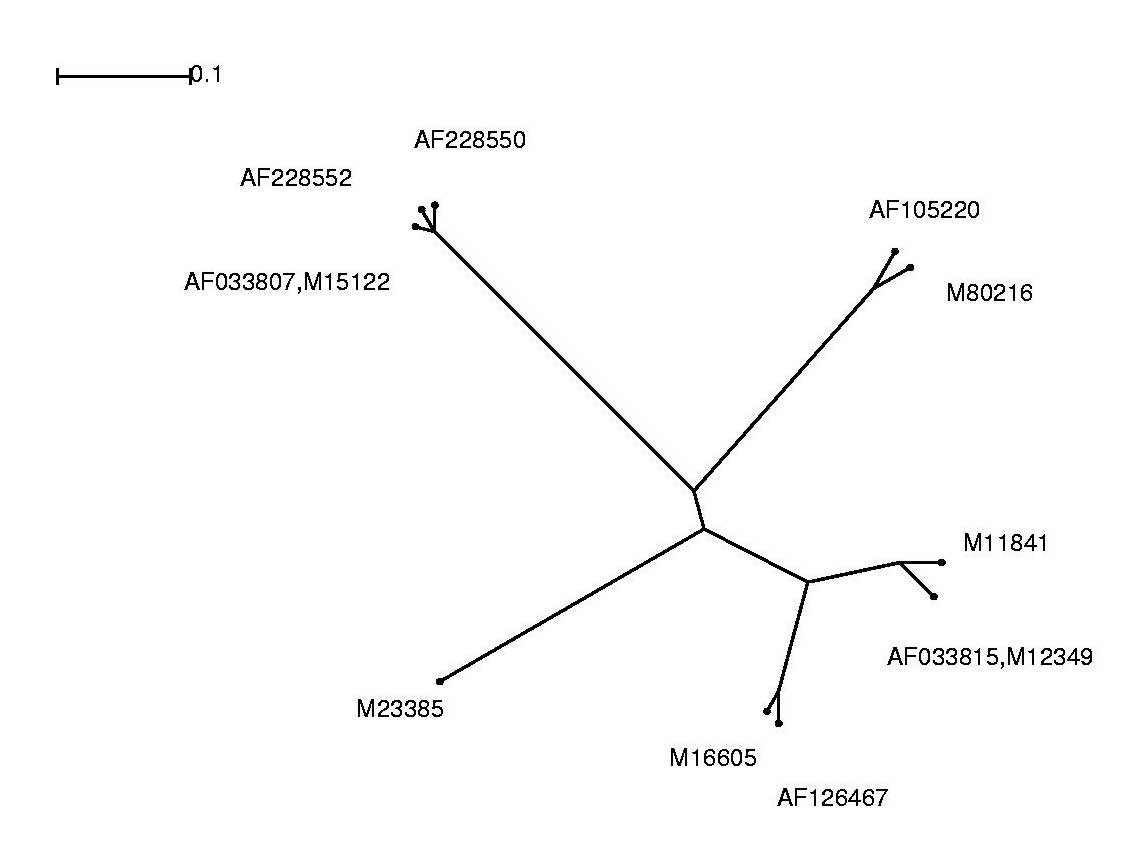
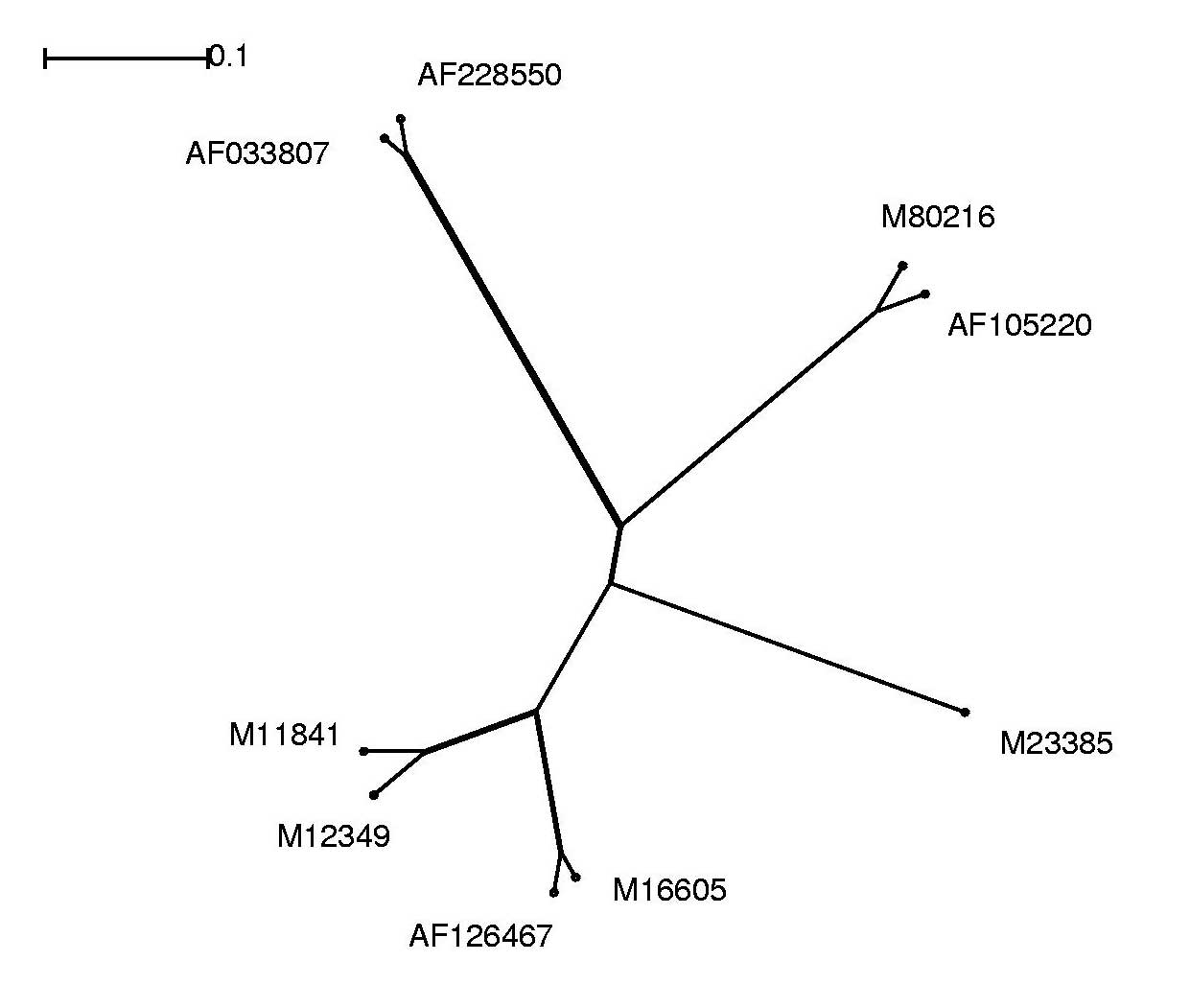
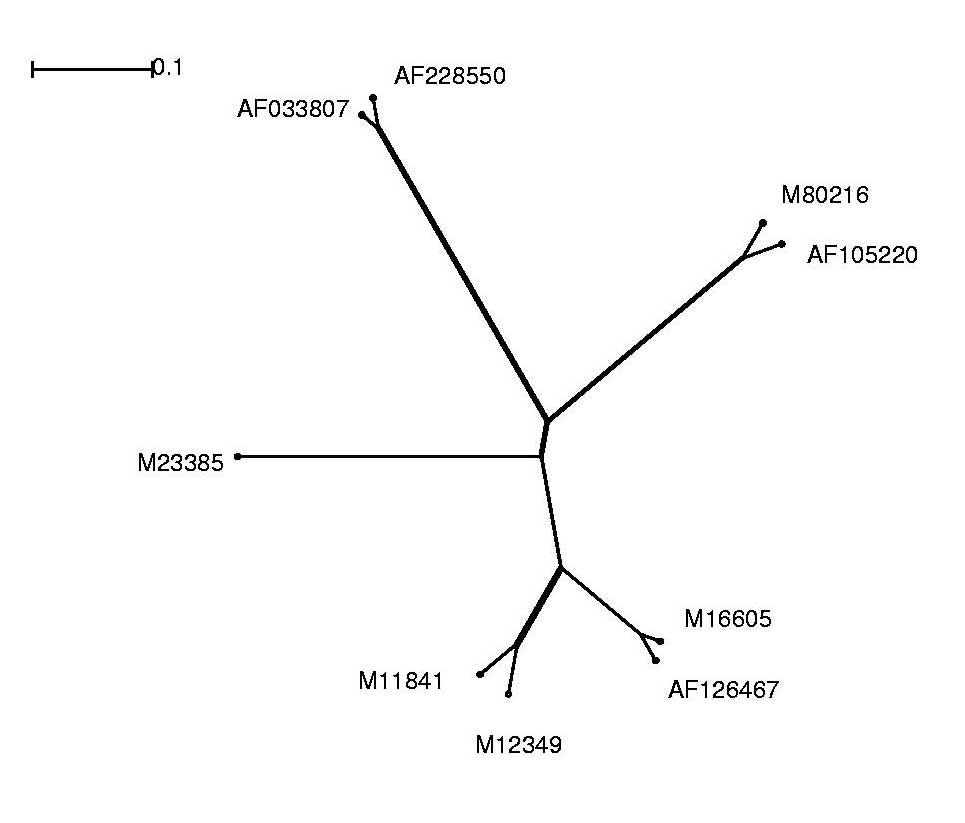
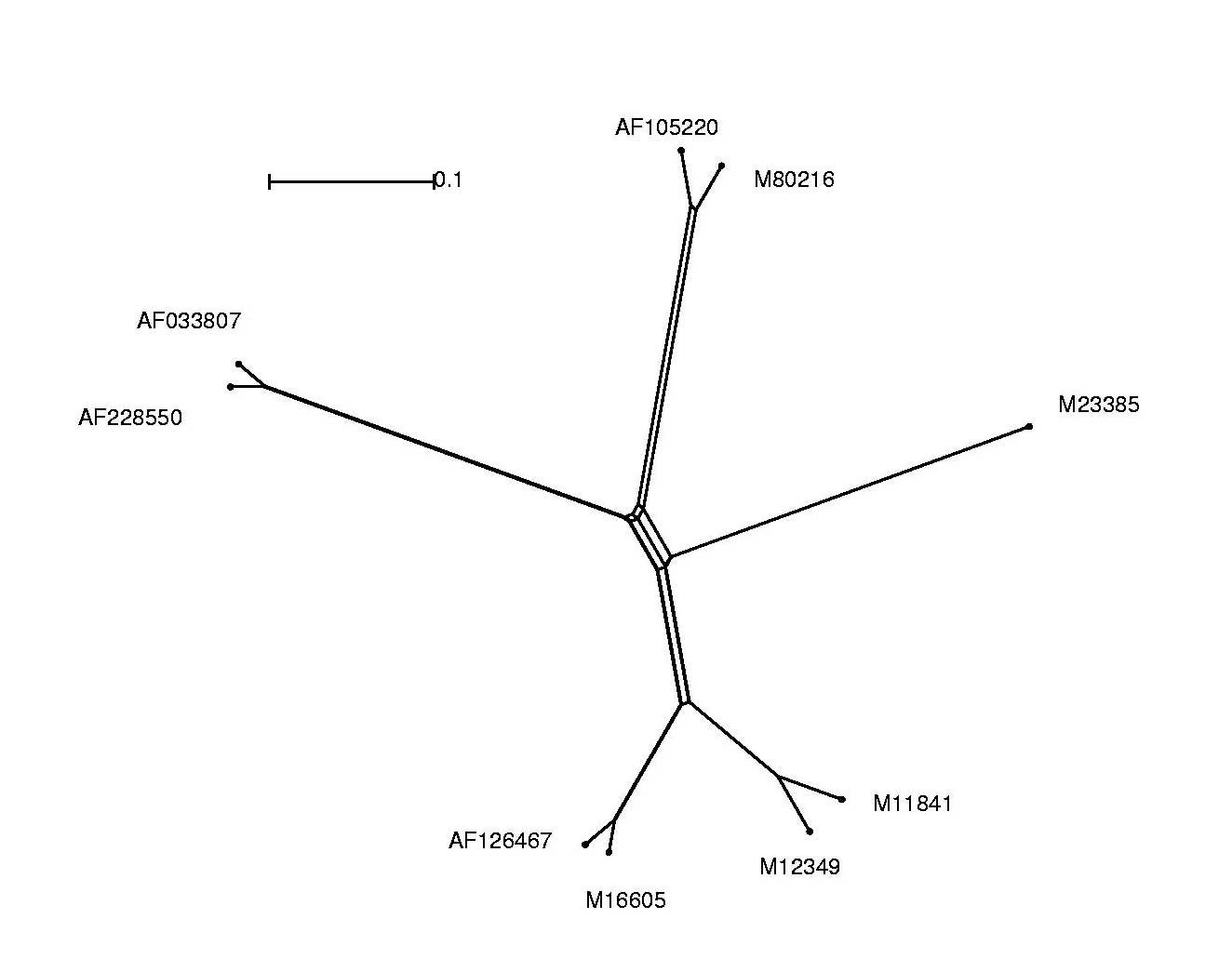
Die Alignmentergebnisse lassen sich als phylogenetischen Baum anschaulicher darstellen und interpretieren.
Hierzu wurde das Programm Splitstree verwendet.
Um die Alignment-Dateien graphisch darstellen zu können, mussten wir das ClustalW-Alignment-Format in das splitstreeeigene Nexus-Dateiformat umwandeln. Dies realisierten wir mit Hilfe eines Perl-Skriptes (aln2nex.pl), das die Konvertierung der Datenformate automatisierte.
Im Folgenden sind die Alignment-Ergebnisse sowohl im Splitstree-Dateiformat, als auch in Form von jpg-Dateien, welche wir aus den original Splitstree Post-Script Ausgaben gewannen, aufgeführt: Anhand der Grafik war leicht zu erkennen, dass wir mit redundanten Sequenzen arbeiteten. Somit entschlossen wir uns für die weitere Analyse folgende Sequenzen auszuschließen und nicht weiter zu betrachten:
Dialign2
An dieser Stelle haben wir entschieden, das es keinen Sinn macht das Alignment mittels Code2aln
weiter zu betrachten, da dieses leider absolut identisch zum ClustalW-Alignment war.
Auf Grund dessen wird das Alignment mit Code2aln von den weiteren Betrachtungen ausgenommen.
Um die Alignment-Dateien graphisch darstellen zu können, mussten wir das ClustalW-Alignment-Format in das splitstreeeigene Nexus-Dateiformat umwandeln. Dies realisierten wir mit Hilfe eines Perl-Skriptes (aln2nex.pl), das die Konvertierung der Datenformate automatisierte.
Im Folgenden sind die Alignment-Ergebnisse sowohl im Splitstree-Dateiformat, als auch in Form von jpg-Dateien, welche wir aus den original Splitstree Post-Script Ausgaben gewannen, aufgeführt: Anhand der Grafik war leicht zu erkennen, dass wir mit redundanten Sequenzen arbeiteten. Somit entschlossen wir uns für die weitere Analyse folgende Sequenzen auszuschließen und nicht weiter zu betrachten:
{kind=link}
| Entfernte Sequenz | Grund |
| AF033815 | Identisch mit M12349 |
| AF228552 | zu große Ähnlichkeit mit AF033807 und AF228550 |
| M15122 | Identisch mit AF033807 |
- 9 Sequenzen
- Code2aln
{kind=link}
{kind=link}
{kind=link}
Step 4 - Analyse
Nun konnten wir uns auf die Suche nach stabilen Sekundärstrukturen begeben.
Wir nutzten das Tool RNAfold des Vienna RNA Package zur Energieberechnung
für die Bildung der RNA-Sekundärstrukturen. Mittels readseq bereiteten wir
unsere FASTA-Sequenzdatei für die Weiterverarbeitung
durch RNAfold auf. Ergebnis dieses Schrittes und Quelldatei für RNAfold war die Datei
data.vienna.
Das rechenintensive RNAfold ließen wir über Nacht auf Cluster-Rechnern verteilt laufen und erhielten das Faltungsergebnis in Form von Ausgabedateien (*.out) bzw. *.ps Dateien. Durch das Perl-Skript split.pl wurde nun für jede Sequenz eine *.mfe Dateie erzeugt.
Dadurch hatten hatten wir alle benötigten Dateien zur Weiterverarbeitung mit Alidot zusammen.
Beide Programme sind Teil des Vienna RNA Package.
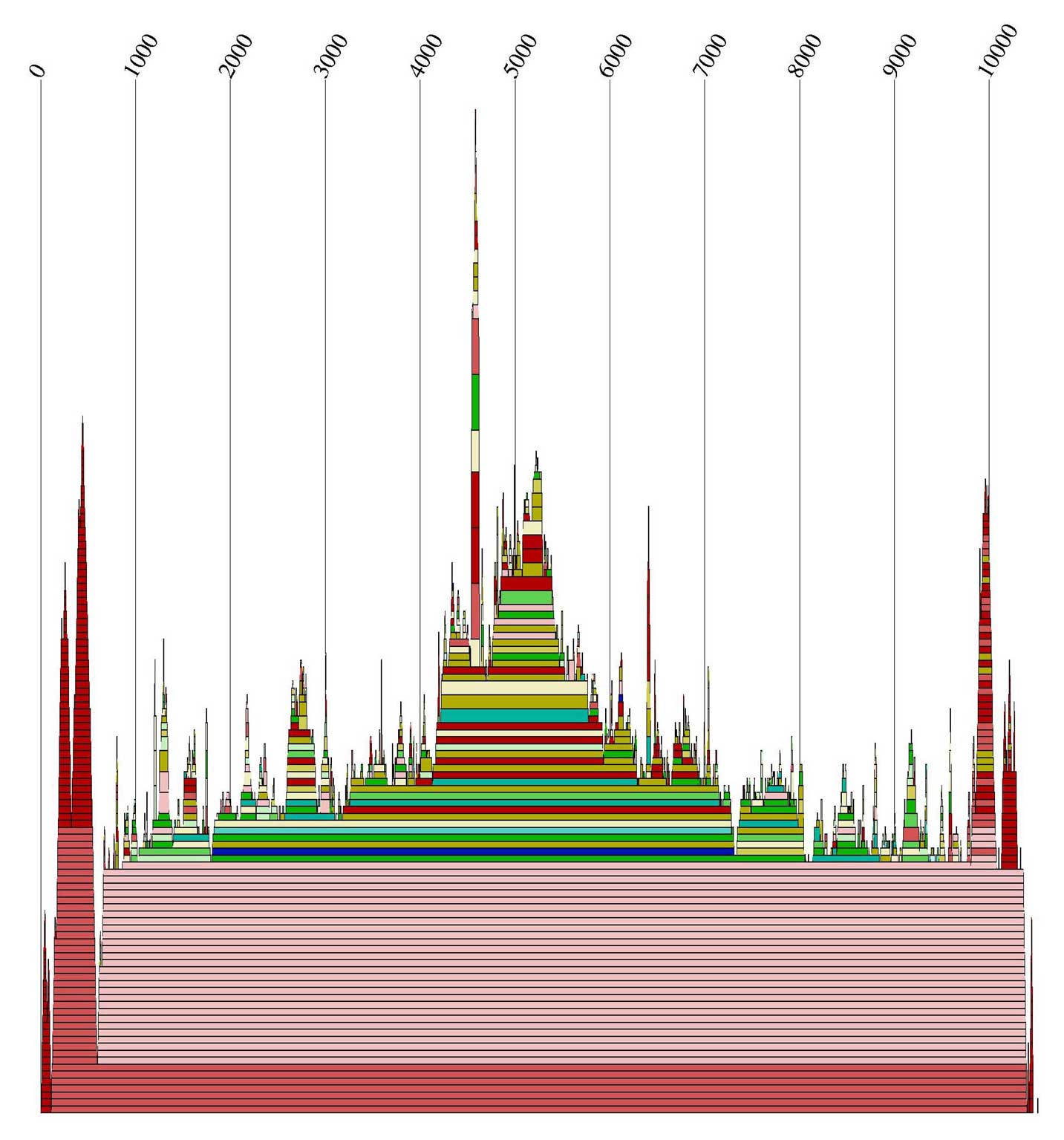
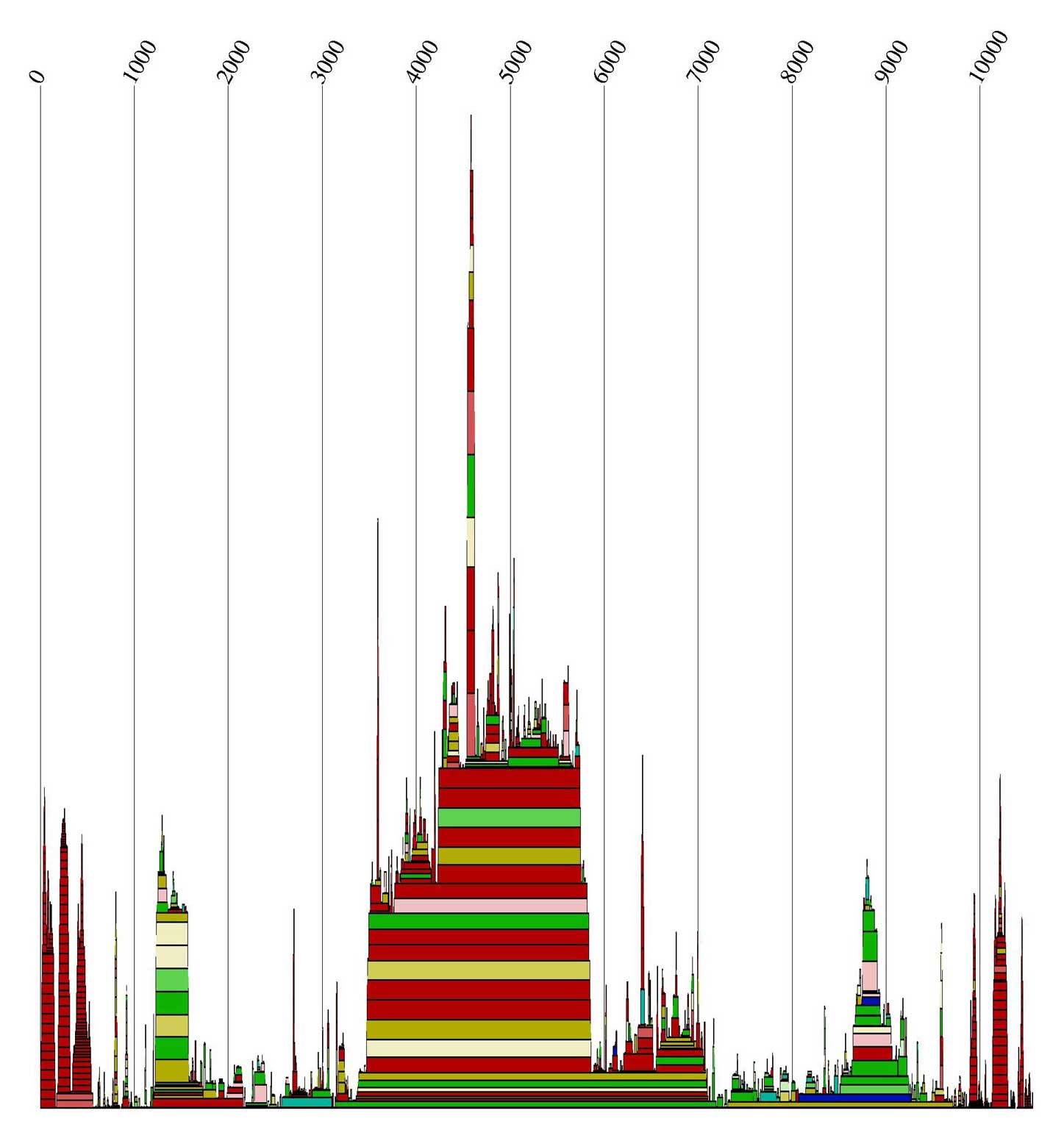
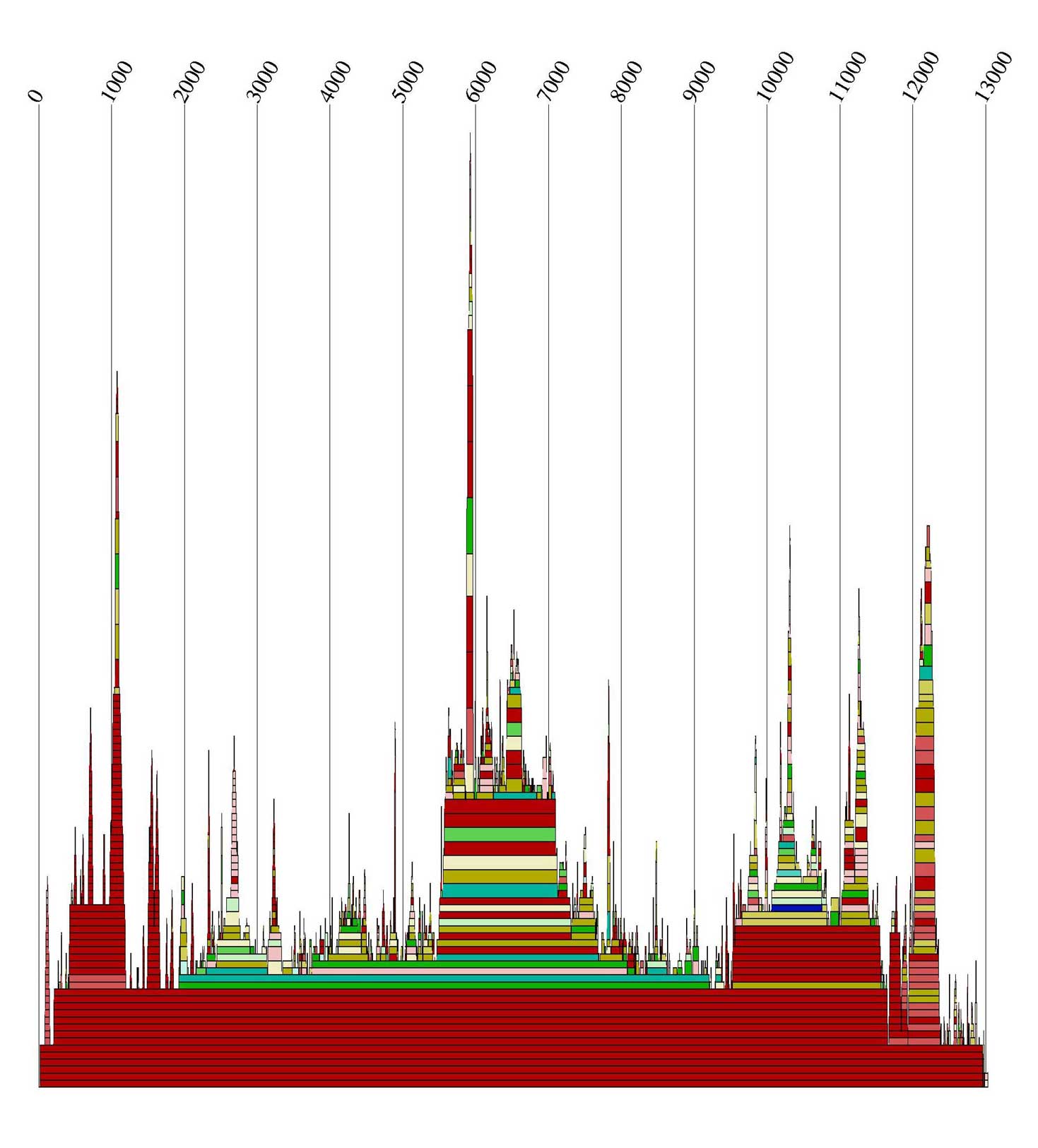
Alidot dient zum Auffinden konservierter Motive. Dazu werden die Informationen über die Einzelsequenzen
zusammengefasst. Wir riefen es auf zwei unterschiedliche Arten auf:
Zum Einen nutzte Alidot die *.mfe Dateien, und zum Anderen benutzte es durch Angabe des Parameters -p
die *.ps Dateien. Die Alidot-Ausgabedateien
enthalten Informationen über die Anzahl und die Wahrscheinlichkeiten, ob eine Basenpaarung vorkommt.
Das rechenintensive RNAfold ließen wir über Nacht auf Cluster-Rechnern verteilt laufen und erhielten das Faltungsergebnis in Form von Ausgabedateien (*.out) bzw. *.ps Dateien. Durch das Perl-Skript split.pl wurde nun für jede Sequenz eine *.mfe Dateie erzeugt.
| Sequenz | RNAfold-Ausgabe-Datei | RNAfold-Dotplot | mfe-Datei |
| AF033807 | AF033807.out | AF033807_dp.ps | AF033807.mfe |
| AF105220 | AF105220.out | AF105220_dp.ps | AF105220.mfe |
| AF126467 | AF126467.out | AF126467_dp.ps | AF126467.mfe |
| AF228550 | AF228550.out | AF228550_dp.ps | AF228550.mfe |
| M11841 | M11841.out | M11841_dp.ps | M11841.mfe |
| M12349 | M12349.out | M12349_dp.ps | M12349.mfe |
| M16605 | M16605.out | M16605_dp.ps | M16605.mfe |
| M23385 | M23385.out | M23385_dp.ps | M23385.mfe |
| M80216 | M80216.out | M80216_dp.ps | M80216.mfe |
- ClustalW:
- Alidot ohne Parameter -p
- Alidot-Ausgabedatei(data.alidot, 866 KB)
- Alidot-Ausgabe-Dotplot(aln_dp.ps, 660 KB)
- Alidot mit Parameter -p
- Alidot-Ausgabedatei(data.alidot.pf, 1782 KB)
- Alidot-Ausgabe-Dotplot(aln_pf_dp.ps, 7400 KB)
- Alidot ohne Parameter -p
- Dialign2:
- Alidot ohne Parameter -p
- Alidot-Ausgabedatei(data.alidot, 866 KB)
- Alidot-Ausgabe-Dotplot(aln_dp.ps, 660 KB)
- Alidot mit Parameter -p
- Alidot-Ausgabedatei(data.alidot.pf, 1782 KB)
- Alidot-Ausgabe-Dotplot(aln_pf_dp.ps, 7400 KB)
- Alidot ohne Parameter -p
- ClustalW
- Dialign2
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Step 5 - Protokoll
Der letzte Schritt des Praktikums war die Erstellung einer geeigneten Dokumentation über alle abgelaufenen Vorgänge.
Damit das Protokoll einfach zugänglich ist, und von so vielen Personen wie möglich ohne zusätzlichen
Software-Aufwand zu betrachten ist, verfassten wir diese Webseiten. Das Ergebnis sehen sie nun vor sich.