


Next: RNA Recognition Motif (RRM)
Up: Bioinformatisches Praktikum
Previous: Bioinformatisches Praktikum
Subsections
Die Basis bildet die Superfamily Datebank in der Version vom 18.01.2009 (964.ass.tab). In der Datenbank wurde nach annotierten Family IDs (siehe Tabelle 1) der hier betrachteten Proteindomänen gesucht. Die Proteine in der SuperFamily werden durch HMM annotiert. Verwendet werden dabei mehrere Modelle für die Familie und die Superfamilie. Domänen werden annotiert wenn mindestens ein HMM einer Familie oder einer Superfamilie einen 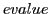 unter einer bestimmten Grenze haben (siehe SuperFamily Webseite für genauere Informationen). Dadurch können Proteine fälschlicherweise annotiert sein, sog. ``false positives''. Aus diesem Grund wurde für jede Proteindomäne ein Subset der gefundenen Proteine erstellt. In dem Subset sind alle Proteine welche mit einem
unter einer bestimmten Grenze haben (siehe SuperFamily Webseite für genauere Informationen). Dadurch können Proteine fälschlicherweise annotiert sein, sog. ``false positives''. Aus diesem Grund wurde für jede Proteindomäne ein Subset der gefundenen Proteine erstellt. In dem Subset sind alle Proteine welche mit einem
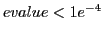 von mindestens einem Family HMM erkannt werden. Für diese signifakanten Subsets sind die Bäume und statistischen Auswertungen verfügbar aber sie werden nachfolgend nicht betrachtet.
von mindestens einem Family HMM erkannt werden. Für diese signifakanten Subsets sind die Bäume und statistischen Auswertungen verfügbar aber sie werden nachfolgend nicht betrachtet.
Table 1:
Proteindomänen und ihre Family ID (ScopID)
| Domäne |
Family ID |
| RRM |
54929 |
| SAM |
47773 |
| PUF |
63611 |
| KH |
54792 |
|
Statistiken
Um zu sehen wie weit die Proteindomänen in bestimmten Taxa verbreitet sind ist es notwendig zu berechnen wie viele Organismen eines Taxon überhaupt in der Superfam bekannt sind. Bei dieser Berechnung, der Anzahl aller Spezies pro Taxon, wurden alle in der Superfamily vorhandenen Spezies auf ihre NCBI ID gemappt. Aus dieser Liste wurden alle mehrfachen Einträge entfernt (da Superfam  NCBI ID nicht injektiv!) da diese im Baum als ein Organismus gezählt werden (bei unterschiedlichen Superfam Organismen mit gleicher NCBI ID wurde derjenige Organismus mit den meisten annotierten Domänen gewählt). Die Organismen wurden dann in dem NCBI-Baum von den Blättern (Organismus) bis zur Wurzel propagiert und in allen durchlaufenen Knoten (Taxa) die Anzahl um eins erhöht. Genauer wurde in einer Hashtabelle jeder Organismus (NCBI ID) mit dem Wert 1 initialisiert und danach für alle Organismen (= Blätter) folgende rekursion angewandt:
NCBI ID nicht injektiv!) da diese im Baum als ein Organismus gezählt werden (bei unterschiedlichen Superfam Organismen mit gleicher NCBI ID wurde derjenige Organismus mit den meisten annotierten Domänen gewählt). Die Organismen wurden dann in dem NCBI-Baum von den Blättern (Organismus) bis zur Wurzel propagiert und in allen durchlaufenen Knoten (Taxa) die Anzahl um eins erhöht. Genauer wurde in einer Hashtabelle jeder Organismus (NCBI ID) mit dem Wert 1 initialisiert und danach für alle Organismen (= Blätter) folgende rekursion angewandt:
- Solange ID
 1 (da NCBI ID 1 die Wurzel ist)
1 (da NCBI ID 1 die Wurzel ist)
- 1.
- ID = NCBI ID des Vaterknotens(ID)
- 2.
- falls Hasheintrag von ID existiert: Hash(ID) += 1
sonst neuer Hasheintrag: Hash(ID) = 1
- 3.
- gehe zu 1
Die so ermittelten Werte repräsentieren also die Anzahl der zugehörigen Organismen eines Taxon.
Die Anzahl annotierter Proteine wurde aus der Datenbank Superfamily gewonnen. Erstaunlich war das die Anzahl annotierter Proteindomänen auch in nahe verwandten Organismen z.T. sehr unterschiedlich war. So wurde z.B. die RRM Domäne in Drosophila melanogaster 335 mal annotiert, in den restlichen Drosophiliden im Schnitt aber viel seltener. Weitere Unterscheide sind in Tabelle 3 zu sehen. Ursache dieser Unterschiede kann die in Abschnitt 1.2 beschriebene Methode der Annotation von Proteindomänen sein. Sind die HMM auf Grundlage dieser Spezies erstellt worden werden in diesen eventuell mehr Treffer gefunden. Aber auch unvollstaendig sequenzierte Genome können eine Rolle spielen (Tabelle 2). Vergleicht man die Annotationen aus dem Subset der signifikanten Proteine wird dieser Unterscheid noch deutlicher (Tabelle 4).
Table 2:
Genomversionen im Vergleich
| Spezies |
Assembly / Release |
| Homo sapiens (Ensembl) |
NCBI36 |
| Pan troglodytes (Ensembl) |
CHIMP2.1 |
| Pongo pygmaeus (Ensembl) |
PPYG2 |
| Mus musculus (Ensembl) |
NCBIM37 |
| Rattus norvgegicus (Ensembl) |
RGSC3.4 |
| Drosophila melanogaster (Flybase) |
5.15 |
| Drosophila yakuba (Flybase) |
1.2 |
| Caenorhabditis elegans (Wormbase) |
Ce147 |
| Caenorhabditis briggsae (Wormbase) |
Cb3 |
|
Table 3:
Unterschiede in der Anzahl annotierter Domänen naher verwandter Spezies.
| |
#Domänen |
| Spezies |
RRM |
SAM |
PUF |
KH |
| Homo sapiens |
825 |
220 |
13 |
111 |
| Pan troglodytes |
578 |
166 |
7 |
73 |
| Pongo pygmaeus |
409 |
104 |
5 |
48 |
| Mus musculus |
786 |
189 |
12 |
84 |
| Rattus norvgegicus |
674 |
163 |
11 |
80 |
| Spermophilus tridecemlineatus |
198 |
58 |
4 |
32 |
| Drosophila melanogaster |
510 |
90 |
7 |
77 |
| Drosophila yakuba |
183 |
41 |
4 |
24 |
| Caenorhabditis elegans |
353 |
46 |
15 |
58 |
| Caenorhabditis briggsae |
171 |
23 |
14 |
29 |
|
Table 4:
Unterschiede in der Anzahl signifikant annotierter Domänen naher verwandter Spezies.
| |
#Domänen |
| Spezies |
RRM |
SAM |
PUF |
KH |
| Homo sapiens |
85 |
16 |
8 |
44 |
| Pan troglodytes |
61 |
11 |
6 |
24 |
| Pongo pygmaeus |
44 |
4 |
3 |
18 |
| Mus musculus |
119 |
14 |
9 |
39 |
| Rattus norvgegicus |
97 |
9 |
6 |
29 |
| Spermophilus tridecemlineatus |
20 |
4 |
2 |
8 |
| Drosophila melanogaster |
51 |
8 |
5 |
1 |
| Drosophila yakuba |
13 |
3 |
1 |
- |
| Caenorhabditis elegans |
31 |
- |
12 |
7 |
| Caenorhabditis briggsae |
12 |
- |
10 |
4 |
|
Es wurden für die Domänen statistische Daten auf den phylogenetischen Baum der NCBI Taxonomie projeziert. Die entstandenen Bäume sind in den ensprechenden Abschnitten zu finden und können dort runtergeladen werden. Die PDF-Version gibt es hier. Die Datei ass_18-Jan-2009.tab.gz kann von der Superfam Webseite bezogen werden. Zum projezieren der Superfam Einträge auf die NCBI wurde diese
Datei erstellt. Eine Liste von allen Knoten im Baum sowie der zugehörigen Anzahl an Organismen im Unterbaum ist in dieser
Datei zu finden.
In Tabelle 5 sind die Skripts, welche im Rahmen des Pratkikums entstanden sind gelistet und kurz erklärt.
Table 5:
Liste von im Praktikum erstellten Perl-Skripts
| Name |
Beschreibung |
| addStats.pl |
Fügt in einen Newick-Baum Daten der statistischen Auswertung ein. Es werden an den Blättern die Werte und an den inneren Knoten jeweils Minimum, Durchschnitt und Maximum dieser Werte des Unterbaumes und die Anzahl darin enthaltener Organismen hinzugefügt. |
| getNCBIname.pl |
Dieses Skript wurde verwendet um die Superfamily DB Organismen auf die NCBI Namen zu projezieren. Als Eingabe wird eine Liste mit den Spalten ``Bezeichner''\t``NCBI ID'' benötigt. Mit diesem Skript wurde die superfam2ncbi.tab erstellt. |
| HowMuch.pl |
Aus einer Liste von NCBI IDs wird anhand des taxonomischen NCBI Baumes eine Liste aus Knoten und zugehöriger Anzahl im Unterbaum enthaltener Organismen (nur diese aus der Eingabeliste) erstellt. Aus der Liste aller in der Superfamily bekannter Organismen wurde mit diesem Skript die Datei viechcount erstellt. |
| kindel.pl |
Zu einem vom Nutzer gewählten Taxon der NCBI (über Name oder ID) werden alle Kind-Knoten des taxonomischen NCBI Baumes und die darin enthaltene Anzahl aus der Superfamily bekannten Organismen ausgegeben. Mit der Option -a werden zusätzlich alle diese Organismen ausgegeben. So kann z.B. die Liste annotierter Organismen in den Metazoa mit den aus der Superfamily bekannten Metazoa verglichen werden. |
| motifpergene.pl |
Join von zwei Listen. Beide Listen haben in der ersten Spalte den ``scientific name'' der NCBI als Schlüssel. In der zweiten Spalte steht entweder die zugehörige Anzahl annotierter Domänen oder annotierter Gene. Diese Werte werden um die Zahl Domänen pro Gen erweitert und in eine neue Liste geschrieben. Das Skript ist Bestandteil der Pipelines. |
| specnames.pl |
Bestandteil der Pipelines. Es joint zwei Tabellen anhand der ersten Spalte. |
| tree2org.pl |
Extrahiert aus einem Newick Baum alle Blattknoten. Wurde verwendet um aus den all.tree Dateien z.B. alle gefundenen Metazoa zu extrahieren (siehe auch kindel.pl und Option -a). Dazu wurde all.tree mit dem Programm ``Dendroscope'' geöffnet, der zu untersuchende Unterbaum komplett markiert und extrahiert (in extra Datei). Diese wurde gespeichert und tree2org.pl auf dem STDIN übergeben. Die so erstellte Liste kann dann für weitere Auswertungen herangezogen werden. |
|
Die Bäume und Dateien wurden wie folgt erstellt.
Für einzelne Domänen (Bsp PUF):
- grep -P ``\t63611\t'' 964.ass.tab
 ass.tab
ass.tab
- ./SIGNI
- ./GETALL (braucht specnames.pl, motifpergene.pl, superfam2ncbi.tab)
- ./GETSIG (braucht specnames.pl, motifpergene.pl, superfam2ncbi.tab)
- tax -f idall.list -n
 all.tree
all.tree
- tax -f idsig.list -n
 sig.tree
sig.tree
- addStats.pl statsall.list all.tree (siehe auch Quellcode)
- addStats.pl statssig.list sig.tree (siehe auch Quellcode)
Für kombinierte Domänen (Bsp. RRM-KH):
- grep -P ``\t54929\t'' 964.ass.tab
 ass1.tab
ass1.tab
- grep -P ``\t54792\t'' 964.ass.tab
 ass2.tab
ass2.tab
- ./JOINIT (braucht specnames.pl, motifpergene.pl, superfam2ncbi.tab)
- tax -f idall.list -n
 all.tree
all.tree
- addStats.pl statsall.list all.tree (siehe auch Quellcode)
Next: RNA Recognition Motif (RRM)
Up: Bioinformatisches Praktikum
Previous: Bioinformatisches Praktikum
root
2009-03-18