Alle der zehn verwendeten Sequenzen wurden zunächst mittels des Alignment-Programmes
ClusAlW alignt. Dabei wurden neben dem multiplen Alignments auch die paarweisen Alignments
durchgeführt. Die Daten aus der Datenbank des NCBI wurden im FASTA-Format gespeichert und konnten
als Eingabe für ClustAlW verwendet werden. Es mussten nur die jeweiligen Header in den FASTA-Dateien
derart angepasst werden, dass die Accession-Number an erster Stelle steht:
Nachdem das geschehen ist, wurden alle Sequenzen, welche in das Alignment aufgenommen werden sollten,
in eine einzige Datei kopiert und das Alignment gestartet. Das Ergebnis der Berechnungen von ClustAlW
ist eine aln-Datei :
3.2 Erzeugung eines phylogenetischen Baumes mit SplitsTree
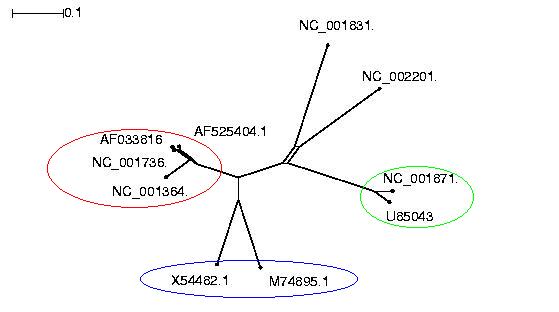
Als nächster Schritt wurde aus der ClustAlW-Ausgabe ein phylogenetischer
Baum erzeugt. Das dafür verwendeten Programm SplitsTree erhält Dateien
im Nexus-Format als Eingabe. Um die aln-Datein in das Nexus-Format zu konvertieren, ist das Perl-Skript
aln2nex.pl benutzt worden. SplitsTree
erzeugte eine PS-Datei des phylogenetischen Baumes:
Bei dem ersten Betrachten des phylogenetischen Baumes fällt auf, dass sich die zehn Sequenzen in drei Gruppen
einteilen lassen, was jeweils engere Verwandtschaft auf Sequenzebene signalisiert. Der entstandene Baum ist
dabei stark von dem Algorithmus abhängig, welcher der Berechnung des phylogenetischen Baumes zugrunde liegt.
3.3 Erzeugung eines phylogenetischen Baumes mit Phylip
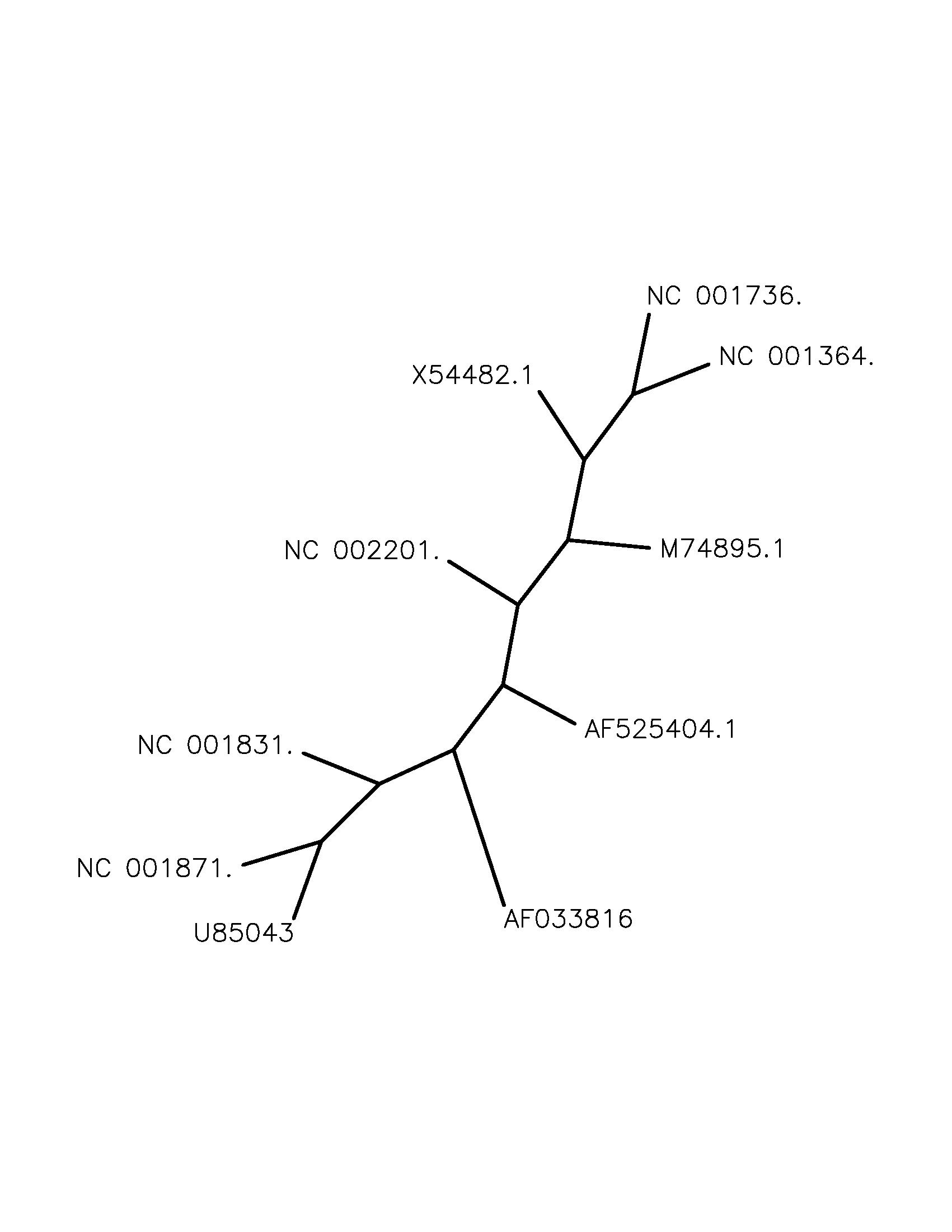
Um einen weiteren Überblick über die phylogenetische Verwandtschaft der Sequenzen zu gewinnen, ist noch
ein zweites Programm zur Erzeugung phylogenetischer Bäume zur Anwendung gekommen. Neben der oben dargestellten
Berechnung von SplitsTree ist das Programm Phylip
verwendet worden. Die phylogenetischen Verwandtschaftverhältnisse der zehn Sequenzen sind abweichend von denen,
die SplitsTree erzeugt hatte. Ausgabe Phylip :
3.4 Erzeugung eines zweiten Alignments mittels code2aln
Zum Vergleich zu den von ClustAlW erzeugten Alignments wurde außerdem noch einmal mit
dem Programm code2alndieser Schritt durchgeführt. Dies diente der genaueren
Untersuchung der Sequenzen, um repräsentative Ergebnisse nicht zu übersehen.
3.5 Berechnung der RNA-Sekundärstrukturen mit RNAfold aus dem Vienna RNA-Package
Die einsträngigen RNA-Moleküle bilden oftmals Wasserstoffbrückenbindungen zwischen paarungsfähigen Basen aus,
so dass Basenpaare entstehen. Dies stellt einen energetisch günsterigen Zustand für das RNA-Molekül dar und
führt zu einer Faltung dessen. Daraus hervorgehende Strukturen sind die schon oft benannten Sekundärstrukturen,
die im Laufe der Evolution auch bestimmte Funktionen für das Virus übernommen haben.
Das im Vienna RNA-Package enthaltene Programm RNAfold wurde an diesem Punkt
verwendet um die Minimum Free Energy (MFE) - Strukturen und die Basenpaarungswatrscheinlichkeiten zu berechnen.
Dazu wurden alle Sequenzdaten zusammen in eine Datei kopiert und das Skript readseq
mit den Optionen: -a -f=19 aufgerufen, um die Sequenzdaten aus dem FASTA-Format in das VIENNA-Format zu konvertieren.
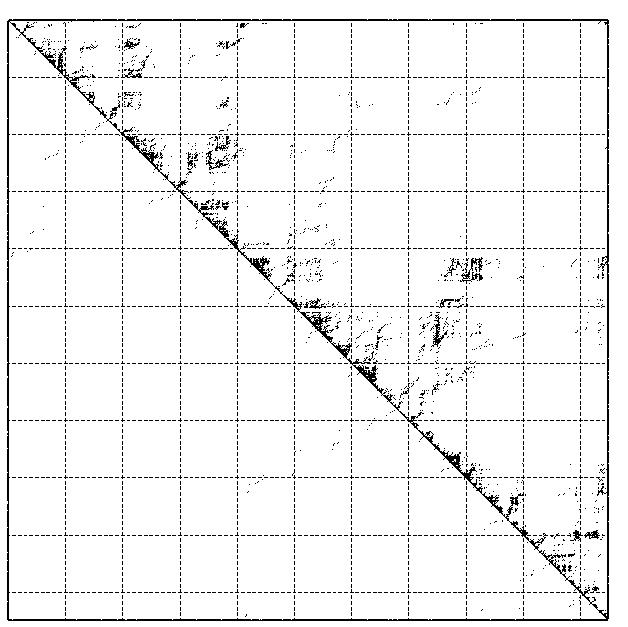
Anschließend wurde das Programm RNAfold mit der Option -p gestartet. Dies
liefert zu den MFE-Daten, welche in Klammernotation zurückgeliefert werden, noch die Basenpaarungswahrschein-lichkeiten, welches
als Dot-Plot erscheinen.
Das Programm alidot dient der Vorhersage konservierter Sekundärstrukturen
des multiplen Sequenz-Alignments.
Dazu wurde alidot mit der Ausgabe des multiplen Alingments von ClustAlW
gestartet. Die Ausgabe wurde in die Datei Alidot.out umgeleitet, in der sich Informationen zu bestimmten Sequenzabschnitten
und der darin enthaltenen Paarungswahrscheinlichkeiten finden lässt.
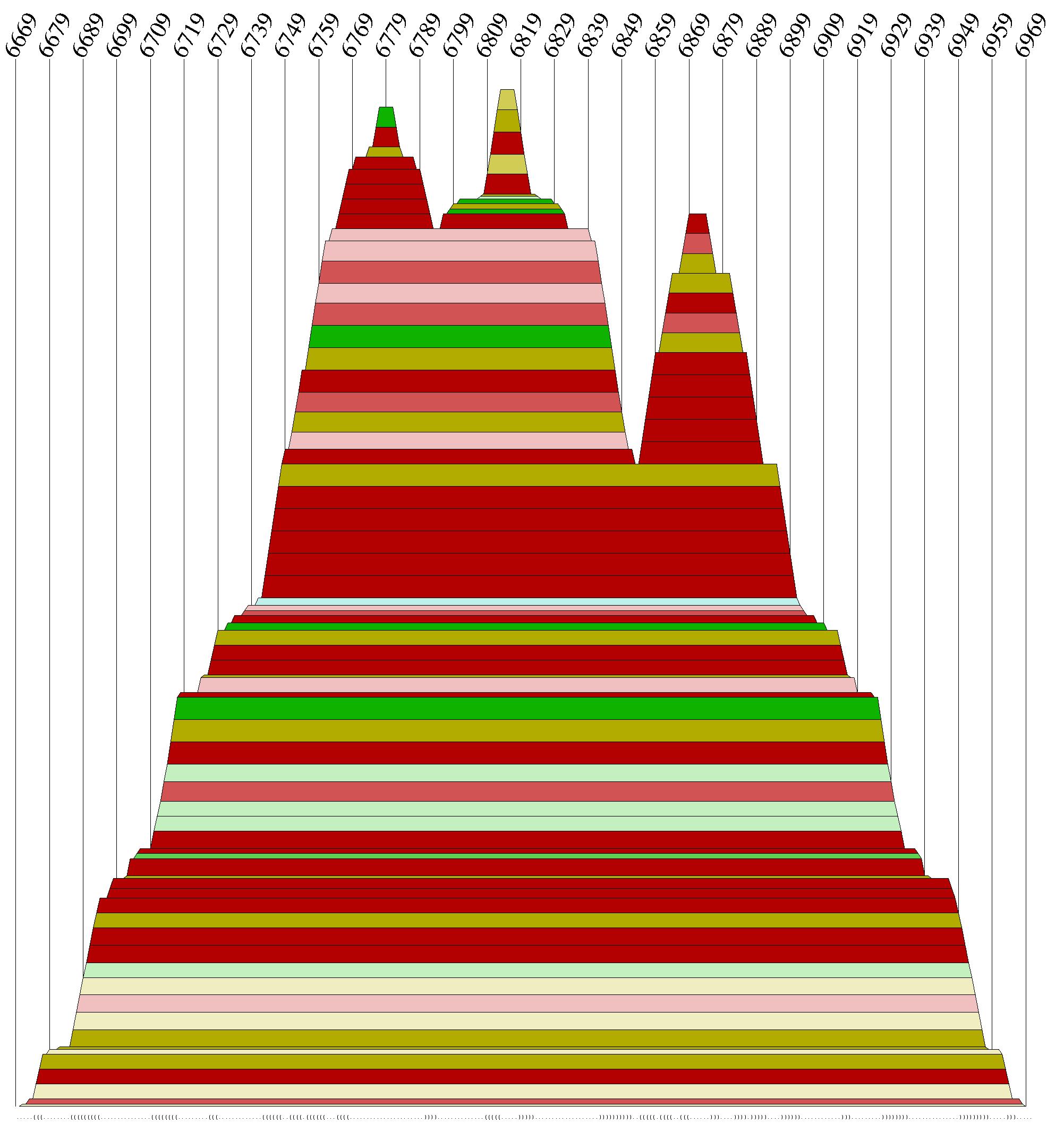
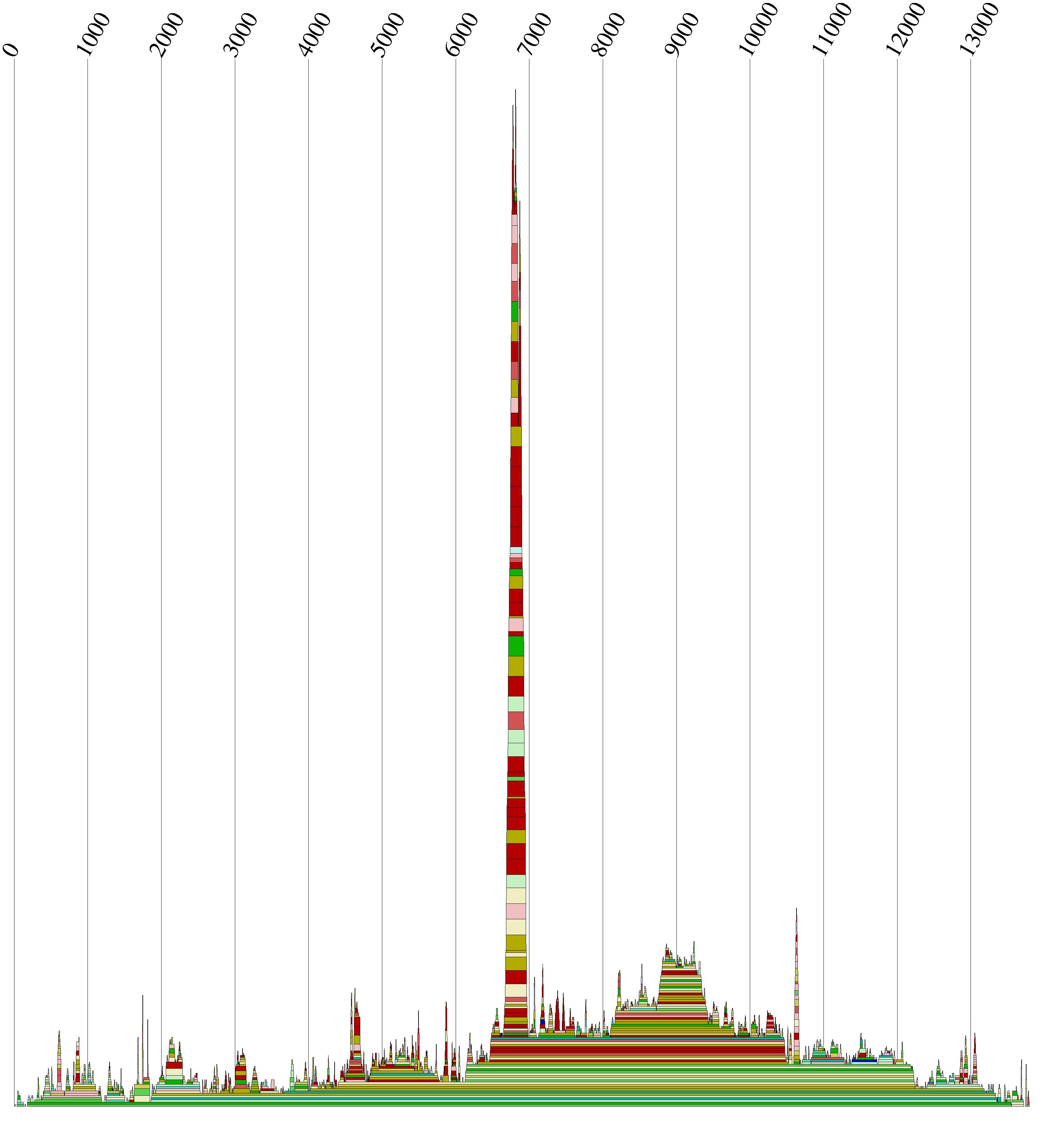
"Diese Datei wurde daraufhin nach Abschnitten geordnet die Paarungswahrscheilichkeiten von mehr als 50%
enthalten. Das Ergebnis waren dann Abschnitte im multiplen Alignment die anschließend als Mountain-Plot
und als 2D-Sekundärstruktur dargestellt worden.
3.7 Darstellung der zu untersuchenden Sequenzabschnitte
Wie schon im vorherigen Abschnitt bschrieben, sind aus alidot-Strukturvorhersage
ein paar Abschnitte im multiplen Alignment entdeckt worden, die besonders hohe Basenpaarungswahrscheinlichkeiten
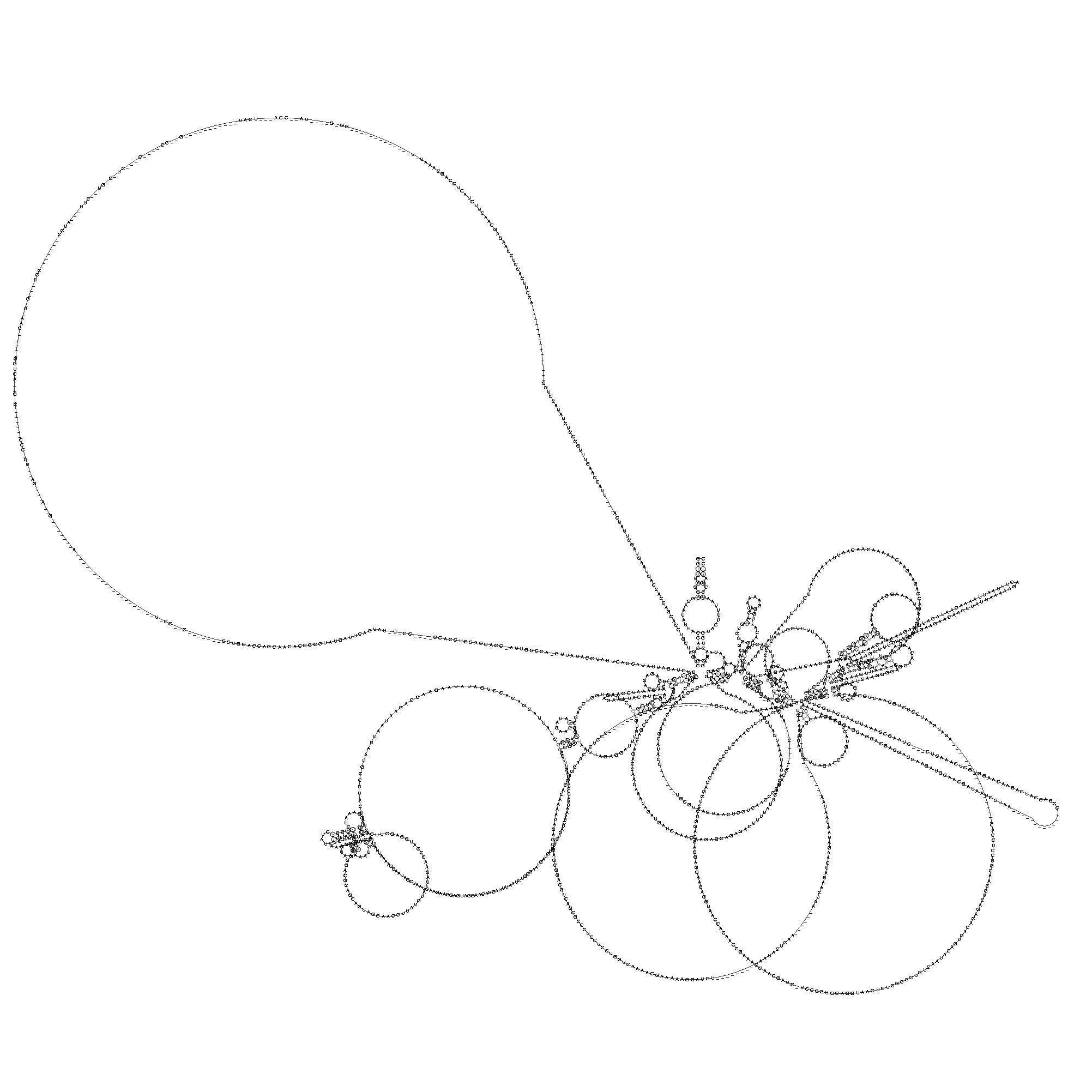
aufwiesen.Diese wurden anschließend verschiedenartig graphisch dargestellt. Mittels RNAplot
wurden die Abschnitte visualisiert. Dazu mussten sie vorher von dem Perl-Skript consensus.pl,
welches die Anfangs- und Endposition sowie die ClustalW-Ausgabe und die alidot-Ausgabe benötigt, extrahiert werden.
Nun konnten Zweidimensionalen Ausgaben erzeugt werden.
Das Perl-Skript dpzoom.pl vergrößert Sequenzausschnitte, so dass sie anschließend
mittels cmount.pl als Mountain-Plot dargestellt werden können. Dazu wird die
von Alidot erstellte Ausgabedatei verwendet und die Anfangs- und Endsequenzposition des entsprechenden Abschnittes
werden als Parameter übergeben.
Auch der Moutain-Plot des gesamten Alignments ist mit cmount.pldurchgeführt worden.