Auswertung
Auswertung
1. Phylogentischer Baum
2. Konservierte Sekundärstrukturen
2.1 Die Sequenz der Basenpaare 4500 - 4800 des Alignments
2.2 Die Sequenz der Basenpaare 1770 - 1840 des Alignments
2.3 Die Sequenz der Basenpaare 6693 - 6989 des Alignments
2.4 Palindromische Sequenzen
1. Phylogenetischer Baum
Bei allen durch die drei verschiedenen Programmen erzeugten phylogenetischen Bäumen ist eine nahezu gleiche Gruppenbildung zu erkennen, auch wenn diese unterschiedlich angeordnet sind.
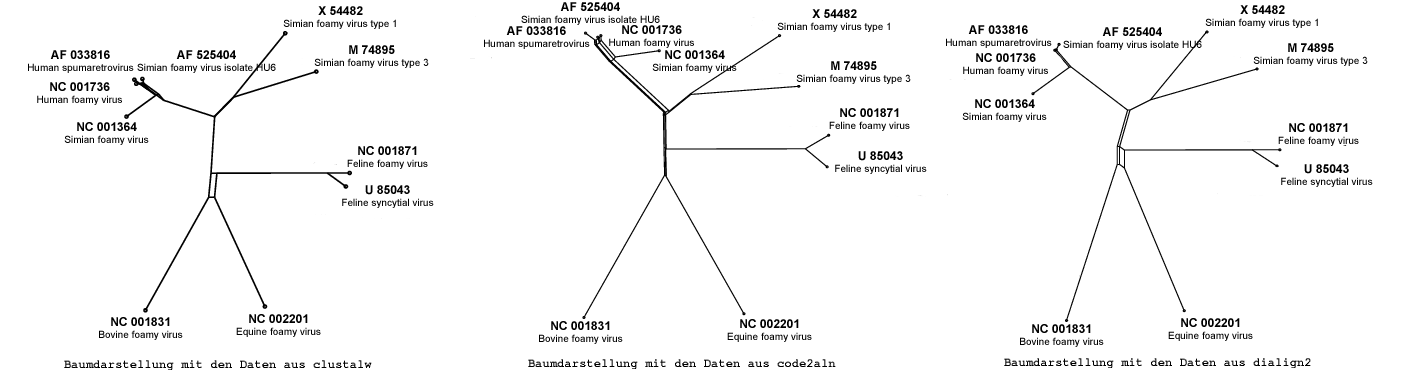
Gruppen bilden zum einen X 54482 und M 74895, beide gehören dem Typ
Simian Foamy Virus an. Eine andere Gruppe besteht aus den beiden
Sequenzen NC 001871 und U 85043, sie zählen zu den Feline-Viren.
Nur schwache Verwandschaft zeigt das Paar NC 01831 und NC 002201, was
sich durch die unterschiedliche Zugehörigkeit erklären lässt,
denn ersterer gehört zu den Bovine und letzterer zu den
Equine-Viren.
Nicht ganz so leicht lässt sich die starke Verwandschaft zwischen den
folgenden 4 Sequenzen AF 525404, AF 033816, NC 001364 und NC 001736
erklären. Zwar gehört sowohl NC 001364 als auch AF 525404 zum
Virus-Typ Simian Foamy, aber ist nicht klar warum sie näher
verwandt zu einer Sequnez des Typs Human foamy Virus und einer des
Typs Human Spumaretrovirus sind als zu den beiden bereits oben
erwähnten Viren ihres Types.
Allerdings ist die Sequenz AF 033816 deutlich kürzer als alle anderen
von uns untersuchten Sequenzen, was unter Umständen ein Grund dieser
Ungereimtheit ist.
| nach oben |
2. Konservierte Sekundärstrukturen
Ausgangspunkt für unsere Untersuchung der Sequenzen auf konservierte Sekundärstrukturen sind der Mountain- und der Dotplot des multiplen Alignments mit allen Sequenzen.

Beim groben Betrachten des Mountainplots des kompletten Alignments fallen einige Regionen auf in denen konservierte Sekundärstrukturen lauern könnten. Besonders sticht dabei der Peak bei 6700-7000 heraus, weshalb wir uns hauptsächlich auf diese Region konzentriert haben. Regionen die wir auch untersucht haben waren 1700-1900, 3600-4000, 4500-4800 und weitere kleine Regionen im Bereich von 5500, 5900 und 12500.
Um weitere geeignete Bereiche für die Suche zu bestimmen, bzw. bereits bestimmte zu bestätigen oder abzulehnen, hilft die Ausgabe der Wahrscheinlichkeiten für konservierte Strukturen des Programms alidot (alidot.out oder ausgabe.txt). Auch hier sticht besonders die Region um 6700-7000 hervor, aber auch die Region 1700-1900 ist gut vertreten.
Um jedoch genauere Aussagen über konservierte Sekundärstrukturen zu machen müssen die Sekundärstrukturen der Abschnitte selber betrachtet werden. Die oben aufgezählten kleineren Regionen sowie die Region 3600-4000 ergeben keine sinnvollen Sekundärstrukturen, sind oft viel zu kurz und von zu geringer Komplexität, weshalb wir diese Regionen für unsere weitere Auswertung außer acht lassen.
Durch Betrachten der einzelnen Regionen und mehrmaliges Herausschneiden und erneutes Alignen der Sequenzen in den jeweiligen Abschnitten, ergaben sich immer exaktere Bereiche. Die von uns gefunden Bereiche in denen wir konservierte Sekundärstrukuren vermuten liegen bei 1770-1840, 4500-4800 und 6693-6989.
Zur besseren Veranschaulichung und Analyse der wahscheinlichen Funktion dieser Sequenzabschnitte wurden diese drei nocheinmal für jeden einzelnen untersuchten Virus farbig auf dessen Sequenz kartiert.
Es wurden folgende Markierungen benutzt:
ocker markiert ist der Bereich 1770 - 1840
violett markiert ist der Bereich 4500 - 4800
blau markiert ist der Bereich 6693 - 6989
| AF 033816, AF 525404, M74895, NC 001364, NC 001736 |
NC 001831, NC 001871, NC 002201, U 85043, X 54402 |

| 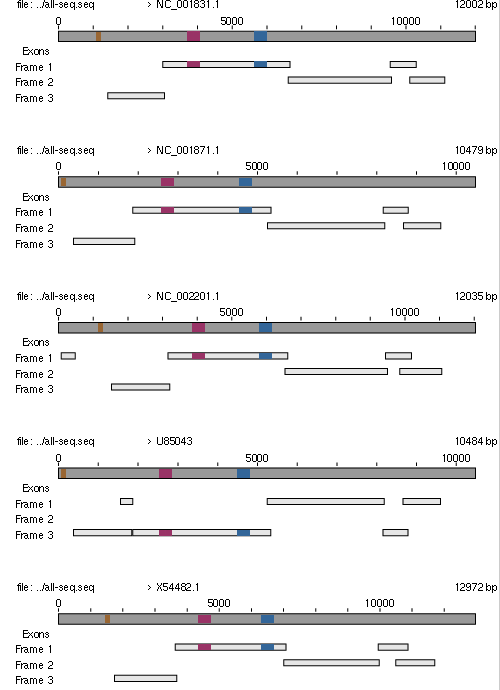 |
Wie bei Retroviren üblich, befindet sich am 5' Ende der RNA-Sequnz das gag-Gen, gefolgt vom pol-Gen und dem env-Gen, welches Information für die Virenhülle in sich birgt
Um den Vermutungen nachzugehen muss jeder Bereich auf Komplexität,
Anzahl der konservierten Basenpaarungen und der Wahrscheinlichkeit dieser hin
untersucht werden.
Zum Abschluss wird der Frage nach der Möglichkeit des tatsächlichen
Einsatzes der Struktur in der Natur nach gegangen und versucht dies mit
Hinweisen aus der Literatur zu bekräftigen.
| nach oben |
2.1 Die Sequenz der Basenpaare 4500 - 4800 des Alignments:
| Sequenzalignment Teil 1 | Sequenzalignment Teil 2 |
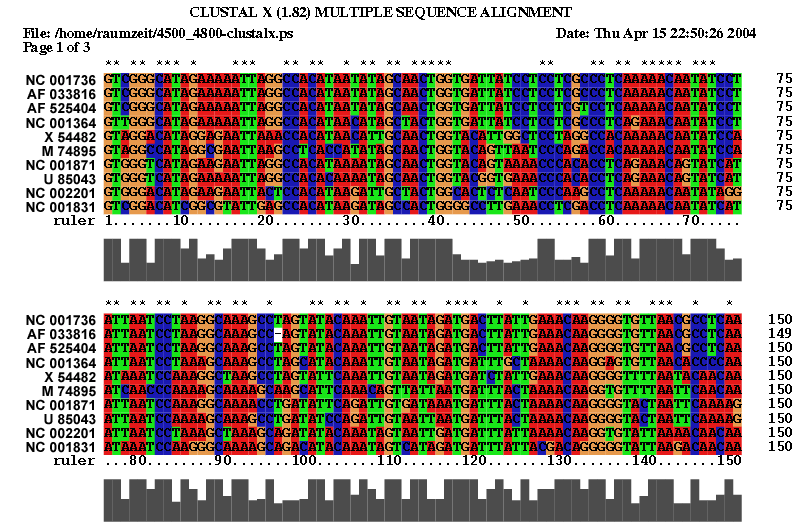 |
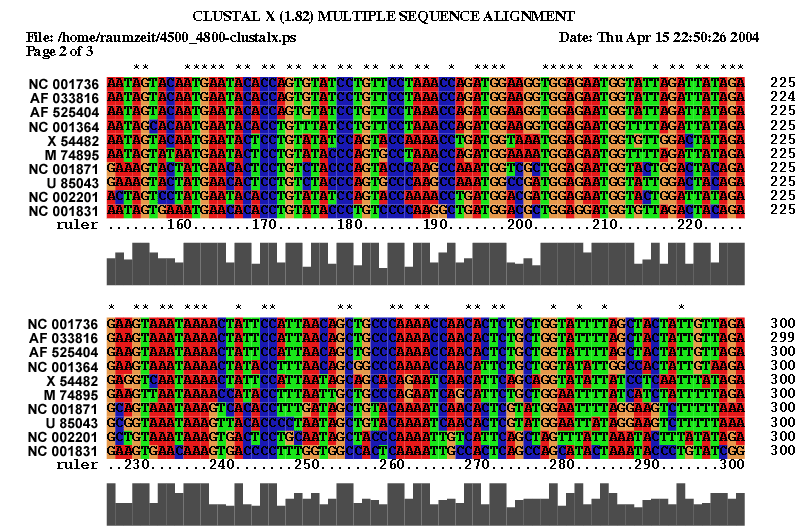 |
| Sequenzalignment Teil 3 | Dot - Plot |
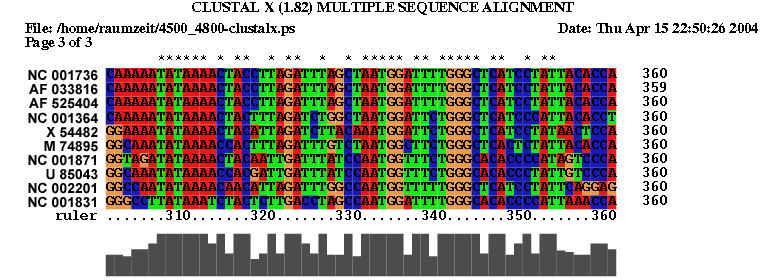 |
 |
| Moutain - Plot | Sekundärstruktur |
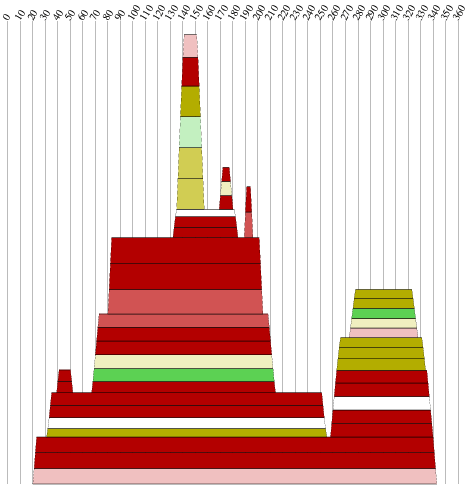 |
 |
Komplexität und Länge der Sequenz sprechen sicher für ein Vorliegen einer konservierten Sekundärstruktur. Allerdings sind nur wenige konservierte Basenpaare, die genügend Wahrscheinlichkeit besitzen vorhanden, hinzu kommt dass auch die Geschlossenheit fehlt. Die Frage nach der Lage der untersuchten Struktur und damit verbunden der Einsatzmöglichkeit, kann nur vage beantwortet werden. Die Region befindet sich bei allen Viren im ORF (Open Reading Frame) des pol-Gens, welches die Informationen für wichtige Proteine wie die reverse Transkriptase, die Protease und die Integrase trägt. Es ist nicht auszuschliessen das die Struktur hilfreich bei der Bildung dieser Proteine ist, allerdings sprechen weder unsere Daten noch Hinweise aus der Literatur dafür. Wir gehen daher nicht von einer hochgradig bedeutenden Struktur aus.
| nach oben |
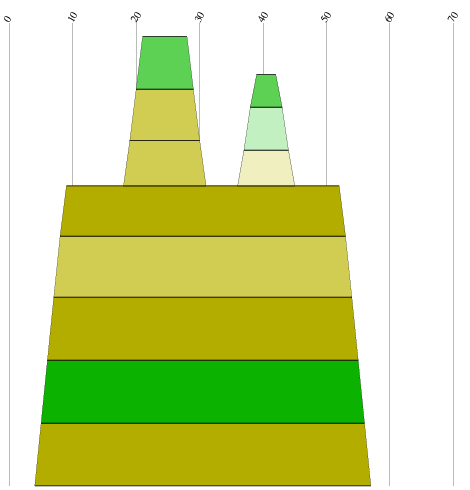
2.2 Die Sequenz der Basenpaare 1770 - 1840 des Alignments:
| Sequenzalignment | Dot - Plot |
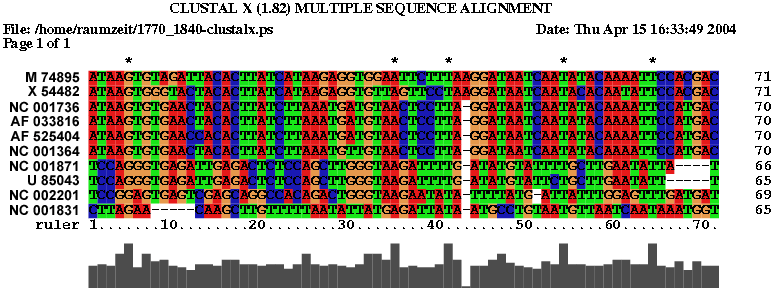 |
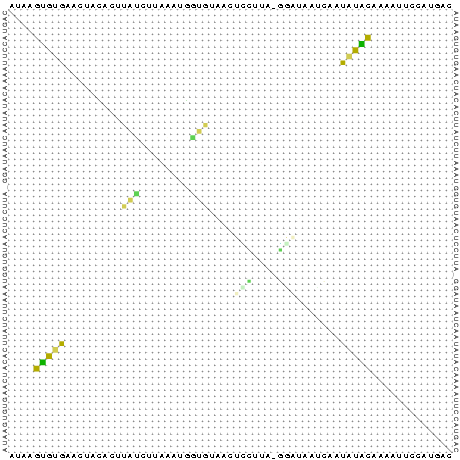 |
| Mountain - Plot | Sekundärstruktur |
 |
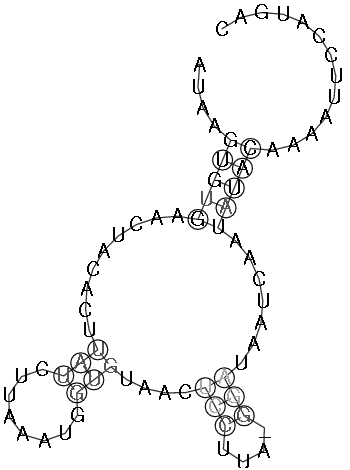 |
Die Struktur besitzt gerade ausreichend viel Komplexität und Länge um nicht
gegen die Überlegung einer konservierten Stelle zu sein. Besonders zu beachten ist, dass
beinahe jede Basenpaarung konserviert zu seien scheint und dies oft auch mit hoher
Wahrscheinlichkeit. Auch die Form der Struktur ist interessant und hält die Hoffnung
auf eine konservierte Sekundärstruktur gestoßen zu sein. Ausschlaggebend wäre
eine eindeutige Aussage über die Lage bzw. den Nutzen der potentiellen konservierten Stelle,
diese kann aber auf Grund fehlender Hinweise aus der Literatur nicht getroffen werden.
Jedoch lassen sich einige Anzeichen finden. Die untersuchte Stelle befindet sich bei fast
allen Viren vor dem Startbereich des ORF des gag-Gens, was bedeuten könnte das sie eine Art
Promotorregion oder eine Bindungsstelle für Ribosomen ist oder eine unterstützende Funktion
für diese darstellt. Jedoch ist ein Zusammenhang mit den palindromischen Sequenzen
nicht auszuschliessen, da sich diese an den von uns untersuchten Sequenzabschnitt anschliessen.
(siehe auch palindromischen Sequenzen)
Es spricht somit viel dafür das es sich hierbei tatsächlich um eine konservierte Sekundärstruktur
der Spumaviren handelt, ob sie weitere Diskussionen und Untersuchungen wert ist überlassen wir dem Leser.
| nach oben |
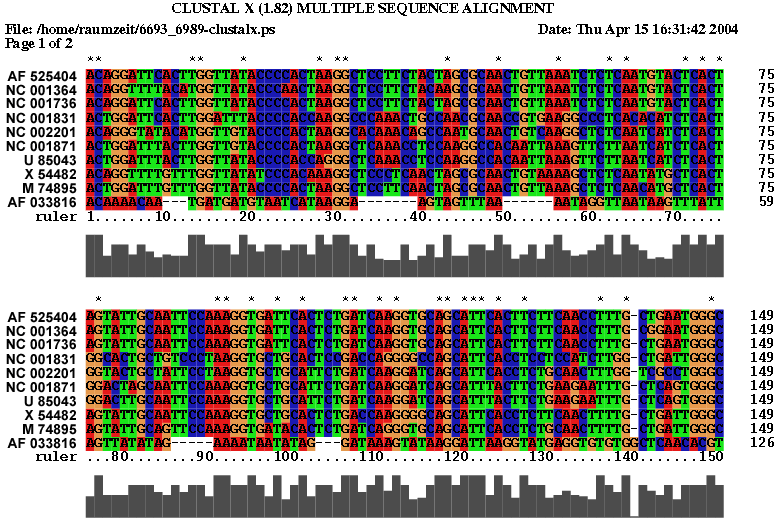
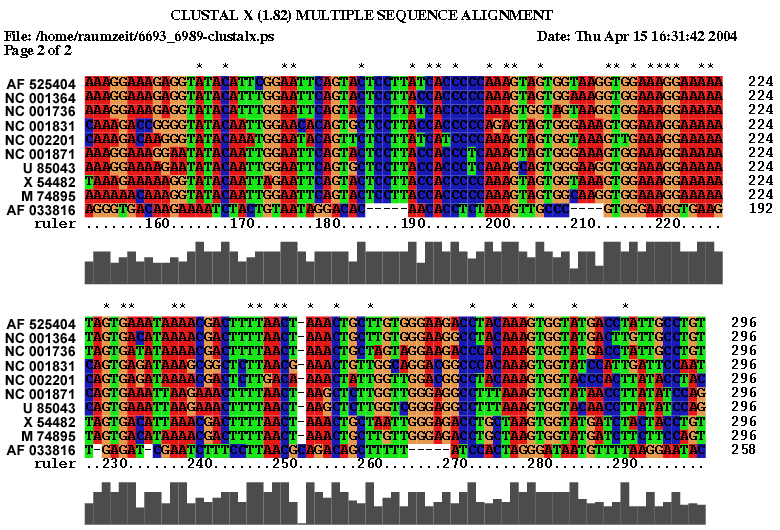
2.3 Die Sequenz der Basenpaare 6693 - 6989 des Alignments:
| Sequenzalignment Teil 1 | Sequenzalignment Teil 2 |
 |
 |
| Dot - Plot | Mountain - Plot |
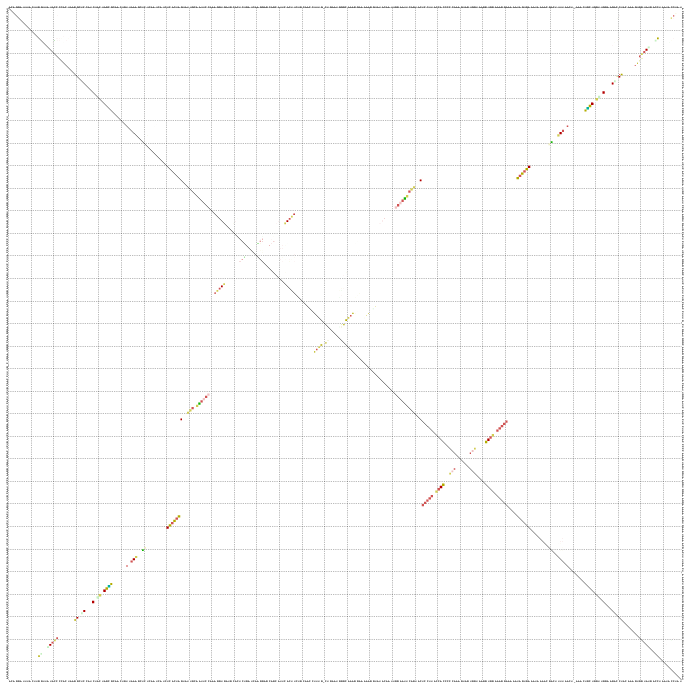 |
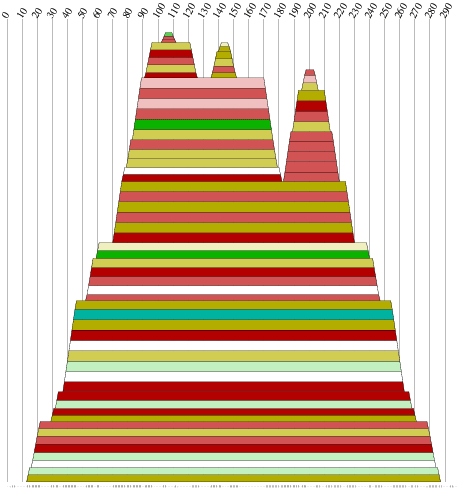 |
| Sekundärstruktur Teil 1 | Sekundärstruktur Teil 2 |
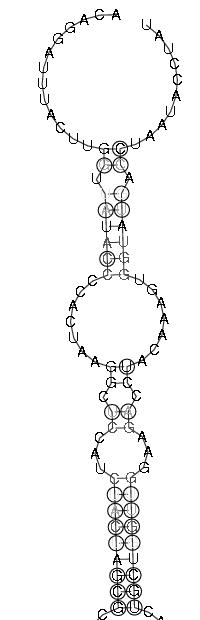 |
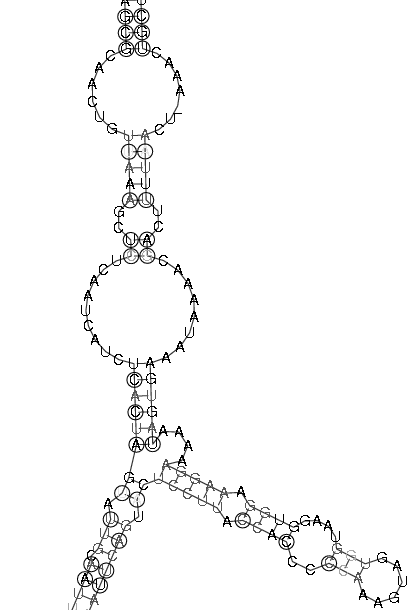 |
| Sekundärstruktur Teil 3 | Sekundärstruktur komplett |
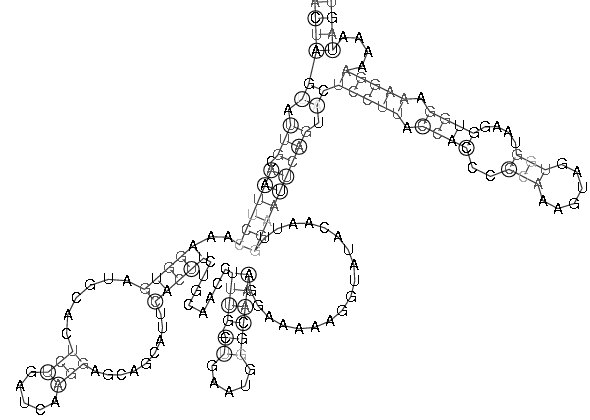 |
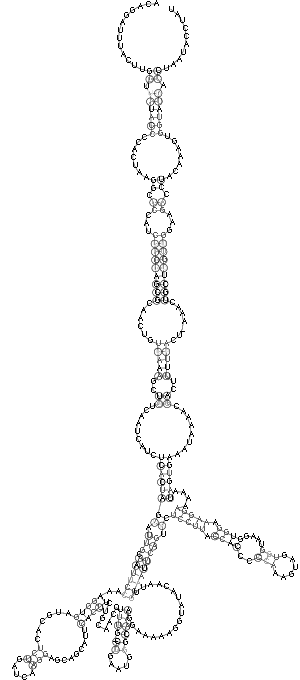 |
Es spricht sehr viel dafür das diese Region eine stark konservierte Struktur ist. Zum einen umfasst sie einen sehr großen Bereich und verfügt über genügend Komplexität um wirklich eine bedeutende Funktion zu besitzen. Zum anderen ist sie gespickt mit konservierten und hoch wahrscheinlichen Basenpaarungen, welche man durch die Kreise um die jeweiligen Basen und die oft schwarze (anstatt graue) Kennzeichnung erkennt. Ein anderer Fakt ist, dass diese Region am 3'-Ende des pol-Gens, also kurz vor dem Beginn des ORF's, welches das env-Gen enthät, beginnt und somit eine hohe Wahrscheinlichkeit besteht, dass die Struktur eine Ribosomenbindungsstelle oder wenigstens einen Promotor darstellt. Obwohl es ohne genaue Kenntnis der tatsächlichen räumlichen Struktur nicht möglich ist, spricht die Form der Sekundärstruktur und ihre daraus ableitbare mögliche Raumstruktur für solch eine Funktion. Dieser Ansatz wird weiter bestärkt durch die bestehende starke Bedeutung des env-Proteins für die Familie der Spuma-viren als einzige innerhalb der Retroviren. Es gibt Hinweise in der Literatur (Links ???), dass es nach Deletion des env-Gens bei Spumaviren nicht zum Austreten der Viren aus einer Zelle und auch nicht zur weiteren Gewebsinfizierung durch Spumaviren kommt. Diese große und bewiesenermaßen, auf Spumaviren beschränkte Bedeutung lässt nur noch den Schluss, dass es sich hier mit höchster Wahrscheinlichkeit um eine hochgradig bedeutsame konservierte Sekundärstruktur dieser Virenfamilie handelt.
| nach oben |
2.4 Palindromsequenzen für die Dimerisierung der doppelt in der Virushülle vorhandenen RNA:
Das Genom der Spumaretroviren liegt in zwei identischen Kopien ihrer RNA, welche
über eine nichtkovalente Bindung in der Nähe des 5' Endes durch eine sogenannte
DLS (dimer linkage sequence) verbunden sind, vor.
Der Mechanismus dieser Dimerisation ist zwar noch nicht ganz geklärt, jedoch gibt es
mindestens zwei Modelle, die diesen erklären könnten. Im kissing-loop-Modell,
welches zuerst für den human immunodeficiency virus type 1 (HIV-1)
vorgeschlagen wurde, geht man davon aus, dass eine palindromische Sequenz, eingebettet
in eine Haarnadelstruktur, der dimer initiation sequence, eine Watson-Crick-Basenpaarung
mit der Palindromischen Sequenz des zweiten RNA-Stranges eingeht, um das Dimer zu formen.
Dieser Mechanismus wurde auch für den avian leukosis sarcoma virus, HIV-2,
simian immunodeficiency virus (SIV) und murine leukemia virus (MLV) propagiert
und scheint ein weit verbreiteter retroviraler Dimerisationsmechanismus zu sein.
Als Alternativmodell gelten purinreiche Motive oder Guaninstrecken, welche wahrscheinlich
durch Bildung von Purinbasen-Tetraedern, stabilisiert durch monovalente Kationen, in den
Dimersiationsprozess involviert sind. Dieses Modell erklärt zum Beispiel die Bedeutung
der guaninreichen Regionen im Moloney murine sarcoma virus, spielt jedoch bei anderen
Retroviren höchstwahrscheinlich keine bedeutsame Rolle und wird deshalb hier nicht betrachtet.
Da die Dimerisation der RNA eine große Rolle im Lebenszyklus des Virus ausmacht,
kann man davon ausgehen, dass die involvierten Sequenzen stark konserviert sind.
Es wurde gezeigt, dass beim HFV (Human Foamy Virus) 3 Sequenzabschnitte für die Dimerisation
eine Rolle spielen (SI, SII, SIII), wovon wir die Palindromische Sequenz SII (UCCCUAGGGA)
in den von uns inspzierten Genomen gesucht haben. Sie befindet sich hochkonserviert an den
folgenden Stellen:
2.4.1
| An der Stelle 541 im Alignment | |
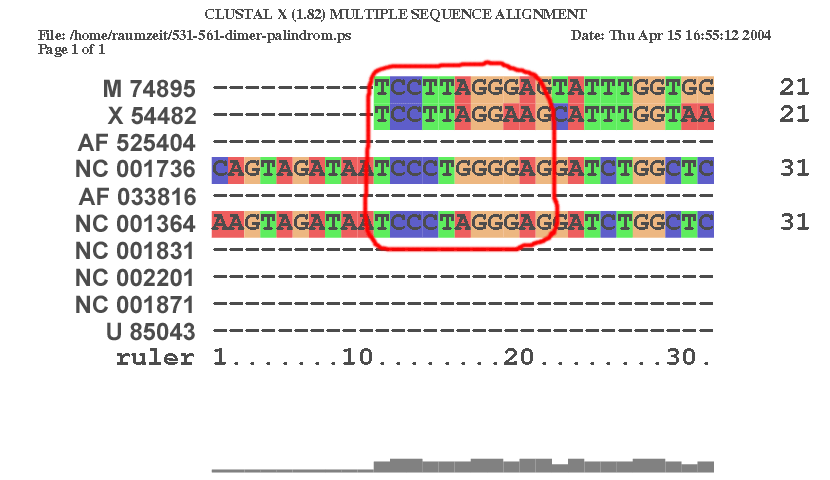 |
|
| Man erkennt deutlich, dass auch M 74895, X 54482 und NC 001736 diese Sequenz einmal besaßen, jedoch ging sie durch Mutation im Laufe der Evolution an dieser Stelle im Genom verloren. | |
| nach oben |
| An der Stelle 1917 im Alignment | |
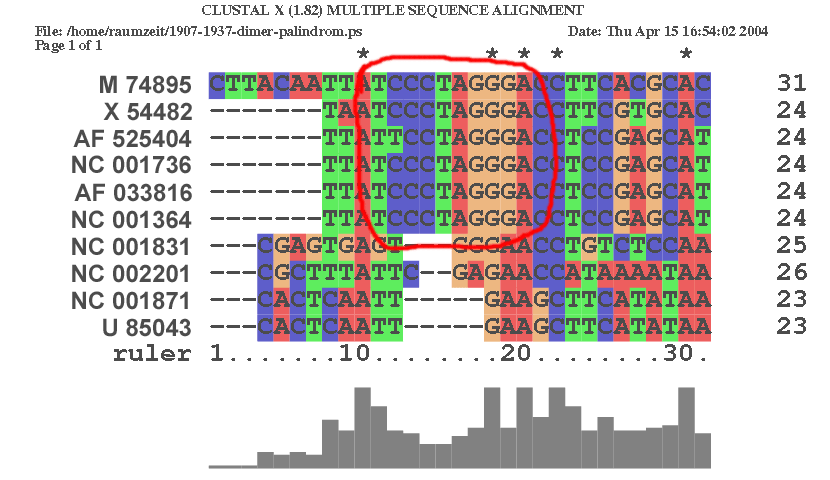 |
|
| Auch hier ist eine Konservierung der palindromischen Sequenz im Laufe der Evolution zu erkennen. AF 525404 zeigt eine Mutaton, welche diese Sequenz für diesen Virus wahrscheinlich unbrauchbar macht. Jedoch sind auch Überreste dieser Sequenz im Genom der anderen Viren auszumachen. | |
| nach oben |
| An der Stelle 2163 im Alignment | |
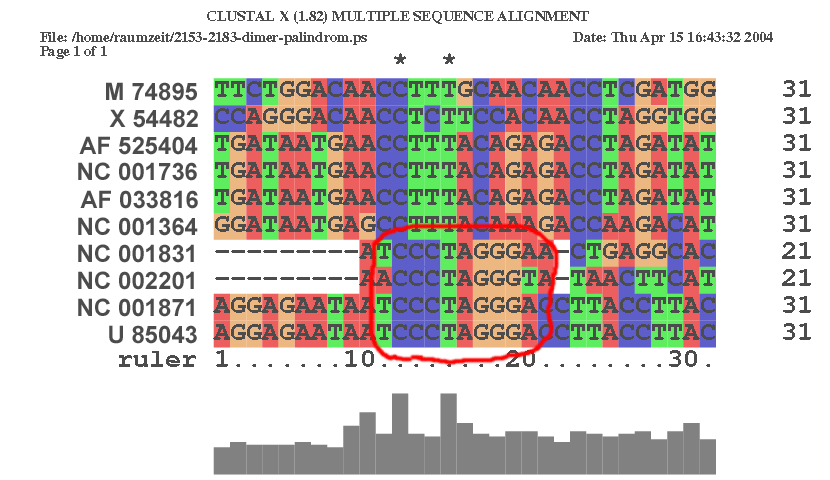 |
|
| An dieser Stelle zeigt sich deutlich die Rolle der konservierten palidromischen Sequenz, da die Sequenz des NC 002201 eine kompensatorische Mutation aufweist. | |
| nach oben |
| An der Stelle 12514 im Alignment | |
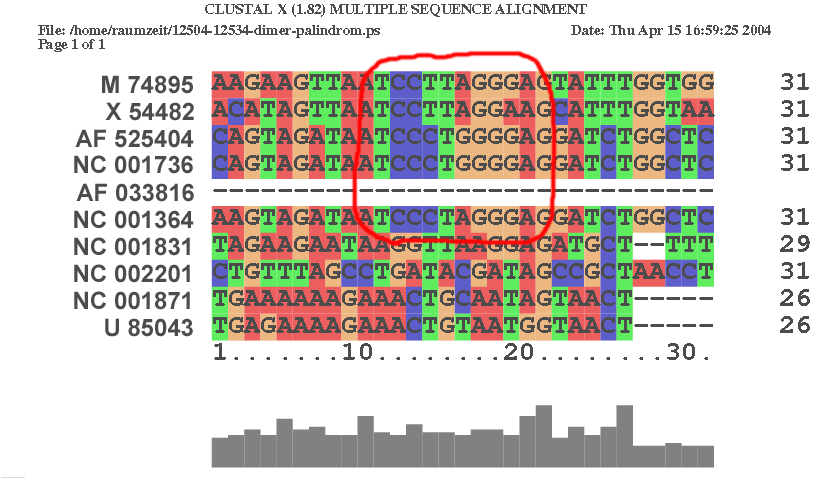 |
|
Interessanterweise befinden sich diese palindromishen Sequenzen downstream von der 1770 - 1840 - Region. Da in der Literatur davon ausgegangen wird, dass die palindromische Sequenz in einer Haarnadel-Struktur enthalten ist, könnte sich so die Funktion der konservierten Struktur an der Stelle 1770-1840 erklären. Eine weitere Untersuchung dieser Region wäre demnach empfehlenswert. Die Ergebnisse der Programme alifold, alidot und cmount für die Stelle 1700 - 2000, welche die 1770 - 1840 - Region als auch die palindromischen Sequenzen enthält, befinden sich in den Ergebnissen der RNA Strukturanalyse.
| nach oben |