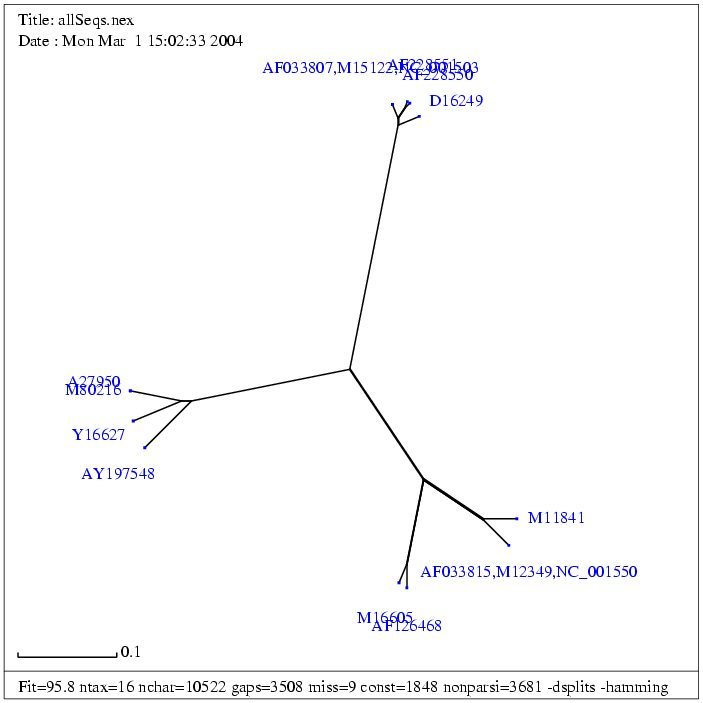
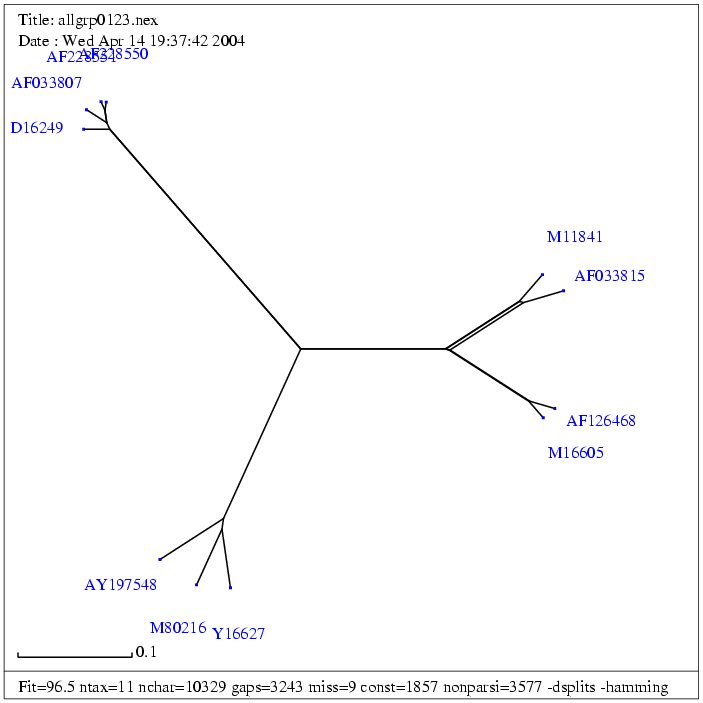
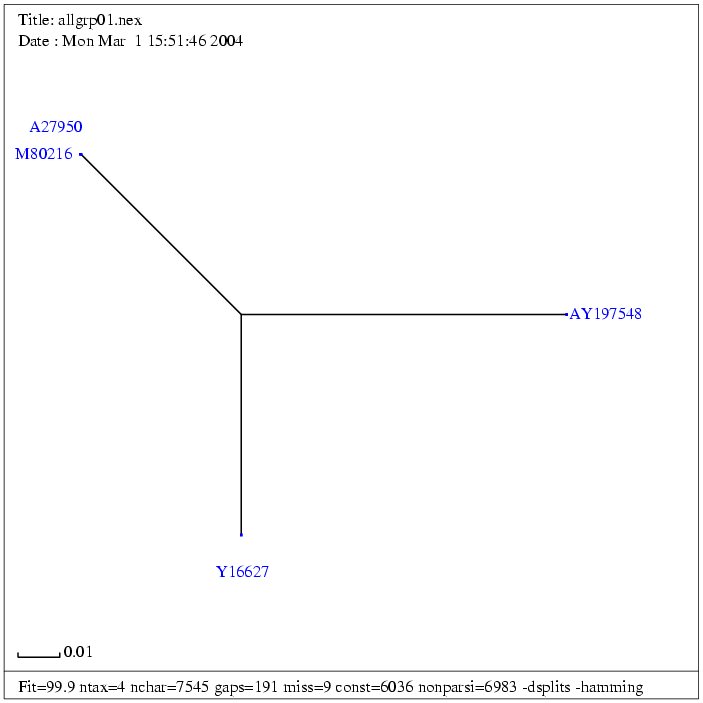
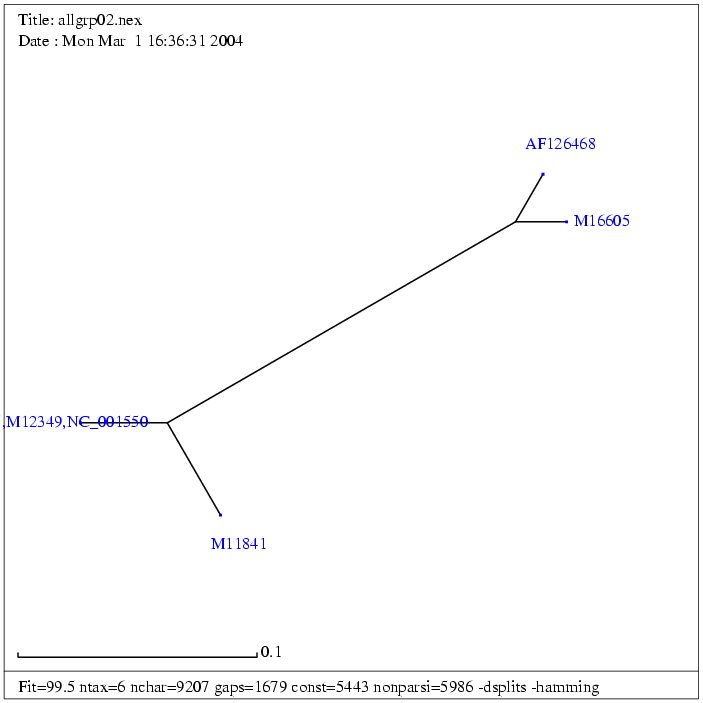
| Gruppe 1 | Fasta |
|---|
| (A27950) | JSRV genomic sequence | 7462 bp | fasta |
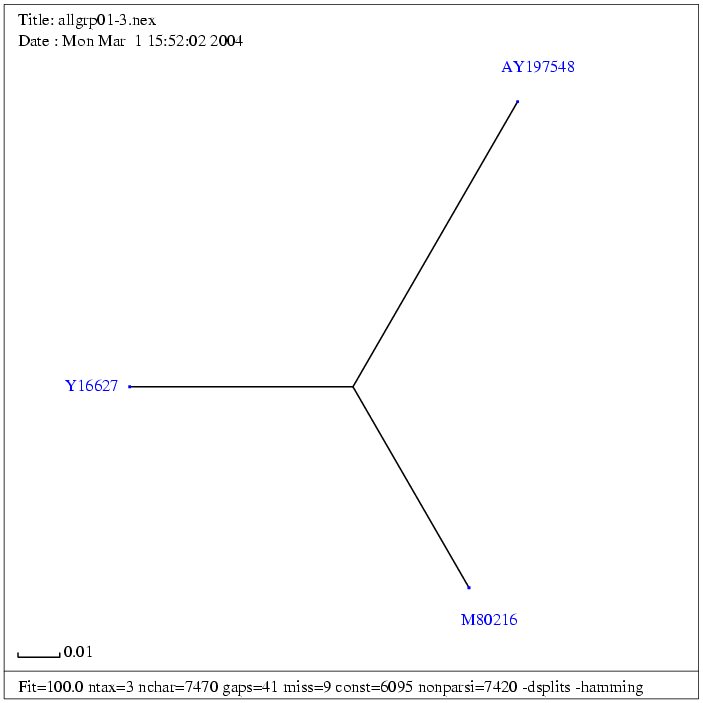
| M80216 | Jaagsiekte sheep retrovirus | 7462 bp | fasta |
| Y16627 | Ovine enzootic nasal tumour virus, complete sequence | 7434 bp | fasta |
| AY197548 | Enzootic nasal tumour virus of goats, complete genome | 7448 bp | fasta |
|
| Gruppe 2 |
|---|
|
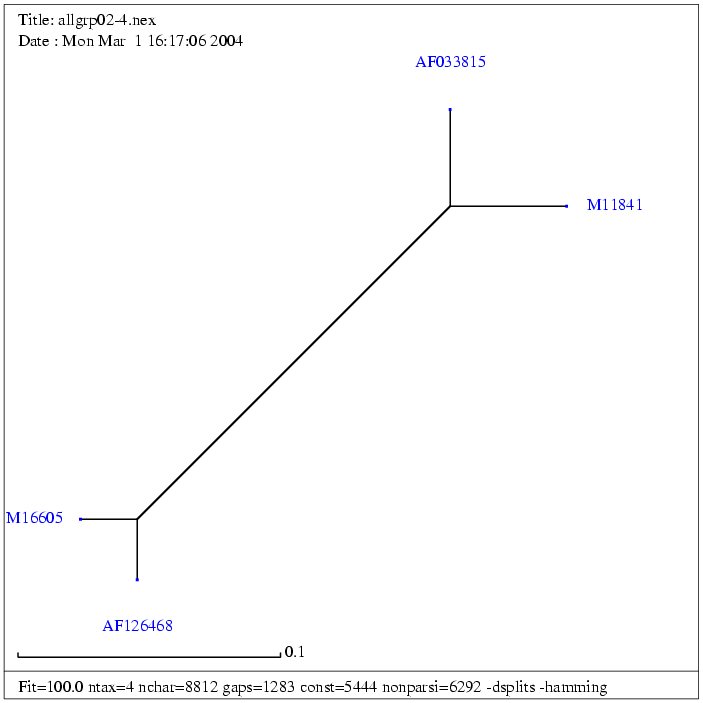
| AF033815 | Mason-Pfizer monkey virus, complete genome | 8557 bp | fasta |
| (NC_001550) | Mason-Pfizer monkey virus | 8557 bp | fasta |
| (M12349) | Mason-Pfizer monkey virus | 8557 bp | fasta |
| M11841 | Simian retrovirus 1 | 8173 bp | fasta |
| AF126468 | Simian retrovirus SRV2 | 8105 bp | fasta |
| M16605 | Simian retrovirus 2 | 7759 bp | fasta |
|
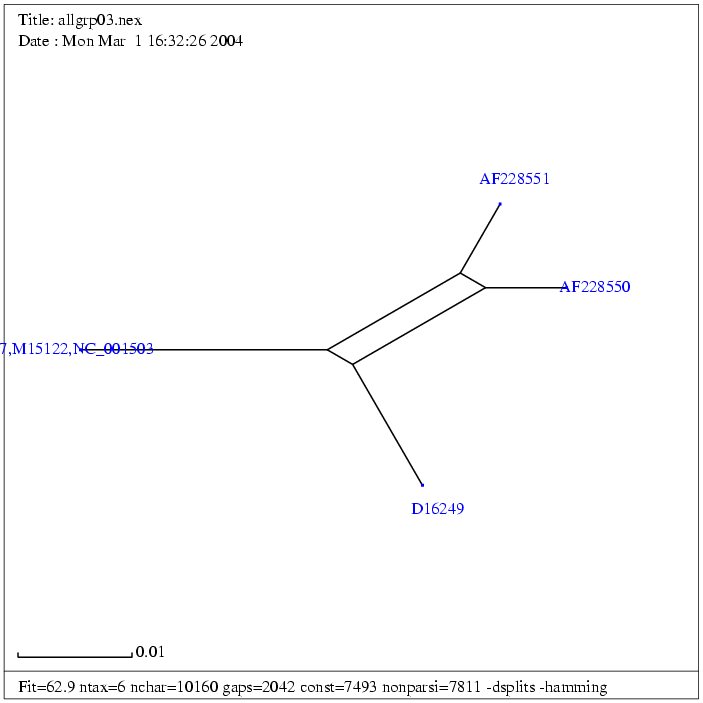
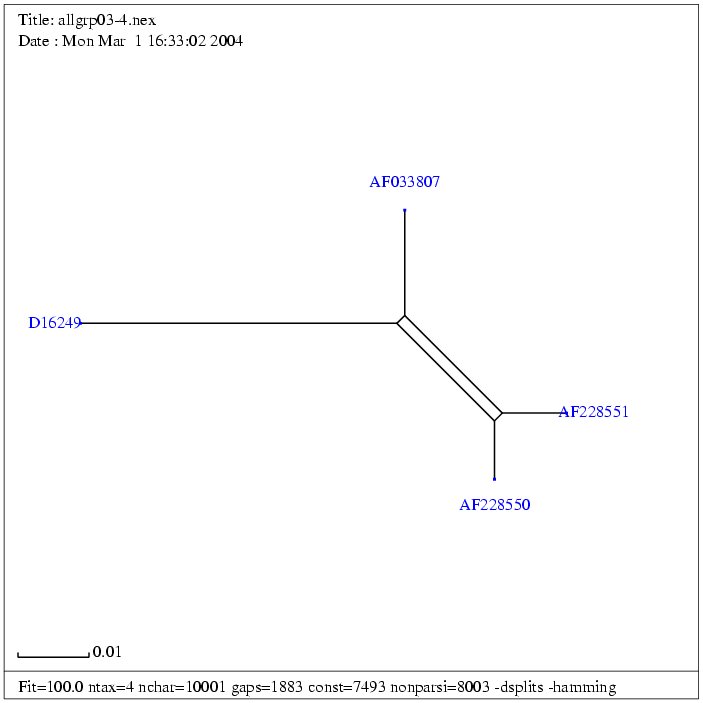
| Gruppe 3 |
|---|
|
| AF033807a | Mouse mammary tumor virus, complete proviral genome | 8805 bp | fasta |
| (NC_001503) | Mouse mammary tumor virus | 8805 bp | fasta |
| M15122b | Mouse mammary tumor virus | 10125 bp | fasta |
| AF228550 | Endogenous mouse mammary tumor virus Mtv1 | 9851 bp | fasta |
| AF228551 | Exogenous mouse mammary tumor virus | 9895 bp | fasta |
| D16249 | Mouse mammary tumor virus proviral DNA
for gag-protease-pol polyprotein and env protein, complete cds4 | 8603 bp | fasta |
Nachdem feststand welche Sequenzen weiter verwendet werden würden, wurden diese Sequenzen mit RNAFold
lokal und auf Rechnern in Wien bearbeitet.
Dieser rechenaufwendige Prozeß nahm einige Zeit in Anspruch.