Erstellung eines Multiple Alignments
Für die zu untersuchende Virusfamilie werden entsprechende Einträge in der NCBI-Datenbank zur Verfügung gestellt. Folgende Viren werden ausgewählt:
| RNA# | Gattung | ID | Länge [nt] | Bezeichnung |
|---|---|---|---|---|
| RNA 1 | Comovirus | AF394608 | 5998 | Bean pod mottle virus strain K-Hopkins1 |
| NC_003496 | 5995 | Bean pod mottle virus | ||
| U70866 | 5995 | Bean pod mottle virus | ||
| M83830 | 5957 | Cowpea severe mosaic virus | ||
| NC_003545 | 5957 | Cowpea severa mosaic virus | ||
| NC_003549 | 5889 | Cowpea mosaic virus | ||
| NC_003741 | 6033 | Red clover mottle virus | ||
| NC_003799 | 5865 | Squash mosaic virus | ||
| Fabavirus | NC_003003 | 5951 | Broad bean wilt virus | |
| NC_003975 | 5957 | Patchouli mild mosaic virus | ||
| Nepovirus | NC_003693 | 7362 | Beet ringspot virus | |
| NC_003509 | 7711 | Blackcurrant reversion virus | ||
| NC_003791 | 7471 | Cycas necrotic stunt virus | ||
| NC_003622 | 7212 | Grapevine chrome mosaic virus | ||
| NC_003615 | 7342 | Grapevine fanleaf virus | ||
| AF016626 | 7977 | Peach rosette mosaic virus | ||
| U50869 | 7514 | Tobacco ringspot virus | ||
| NC_004439 | 7358 | Tomato black ring virus | ||
| NC_003840 | 8214 | Tomato ringspot virus | ||
| RNA 2 | Comovirus | AF394609 | 3688 | Bean pod mottle virus strain K-Hopkins1 |
| NC_003495 | 3662 | Bean pod mottle virus | ||
| NC_003544 | 3732 | Cowpea severe mosaic virus | ||
| NC_003550 | 3481 | Cowpea mosaic virus | ||
| NC_003738 | 3543 | Red clover mottle virus | ||
| AF059532 | 3369 | Squash mosaic virus strain Arizona | ||
| AF059533 | 3356 | Squash mosaic virus strain Kimble | ||
| NC_003800 | 3354 | Squash mosaic virus | ||
| Fabavirus | NC_003004 | 3607 | Broad bean wilt virus | |
| NC_003974 | 3591 | Patchouli mild mosaic virus | ||
| Nepovirus | AY017339 | 3820 | Arabis mosaic virus | |
| NC_003694 | 4662 | Beet ringspot virus | ||
| NC_003502 | 6405 | Blackcurrant reversion virus | ||
| NC_003792 | 4669 | Cycas necrotic stunt virus | ||
| NC_003621 | 4441 | Grapevine chrome mosaic virus | ||
| NC_003623 | 3774 | Grapevine fanleaf virus | ||
| NC_004440 | 4633 | Tomato black ring virus | ||
| NC_003839 | 7273 | Tomato ringspot virus |
Zunächst wird mit dem Programm
clustalw das gemeinsame
Multiple Alignment der ausgewählten Sequenzen der Virusgattungen Comovirus,
Fabavirus und Nepovirus erstellt. Die Alignments werden getrennt für die
RNA-Typen 1 und 2 erstellt.
Hier kann das Alignment von RNA 1 und
RNA 2 betrachtet werden.
Auswertung der Verwandschaftsbäume
Comoviridae
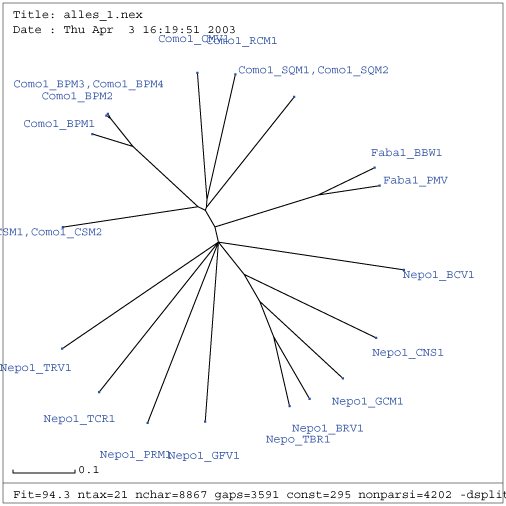
Verwandtschaftsbäume stellen die evolutionären Distanzen zwischen verschiedenen Arten grafisch dar. Der auf Basis des ersten Multiple Alignment mit splitstree berechnete Verwandtschaftsbaum zeigt folgendes Ergebnis.
| RNA 1 | RNA 2 |
|---|---|
|

|
| Verwandtschaftsbaum RNA 1 | Verwandtschaftsbaum RNA 2 |
Hierfür musste zuerst das mit clustalw gewonnene Alignment mit dem Perl-Script aln2nex.pl in das NEX-Format konvertiert werden.
Die Bäume stellen die Hamming-Distanzen der untersuchten Sequenzen dar. In beiden Abbildungen ist eine Aufteilung des Baumes in drei Segmente näherer Verwandtschaft der Viren zu erkennen. Jeder Bereich kann einer Virusgattung zugeordnet werden.
Die Eliminierung nah verwandter Virusarten erhöht die Differenzierbarkeit
einzelner Virenarten im Baum. Schranke der Eliminierung ist der Score des Alignments
der Viren-RNAs. Der Score kann als Verwandtschaftsgrad interpretiert werden.
Sequenzen, deren Score sich im Bereich zwischen 30% und 90%
bewegt bleiben erhalten.
Für die Gattungen Comovirus, Fabavirus und Nepovirus wird nach der
Erstellung eines entsprechenden Alignments ein separater Baum generiert.
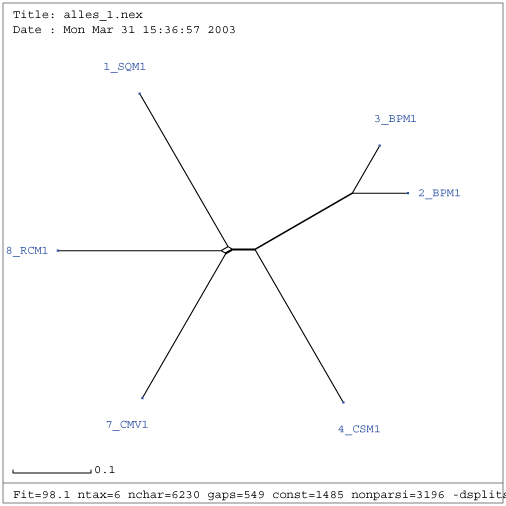
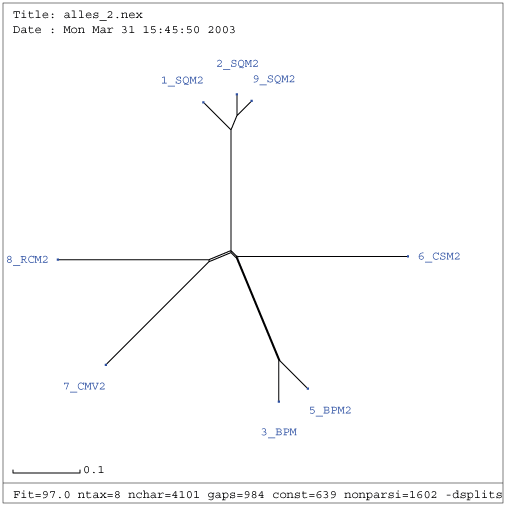
Gattung Comovirus
| RNA 1 | RNA 2 |
|---|---|
|
|
| Verwandtschaftsbaum RNA 1 nach Entfernung der Viren NC_003496, U70866 und M83830 | Verwandtschaftsbaum RNA 2 nach Entfernung des Virus AF394609 |
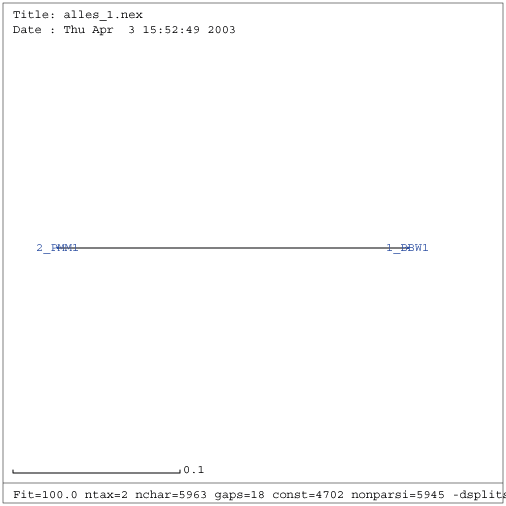
Gattung Fabavirus
| RNA 1 | RNA 2 |
|---|---|
|
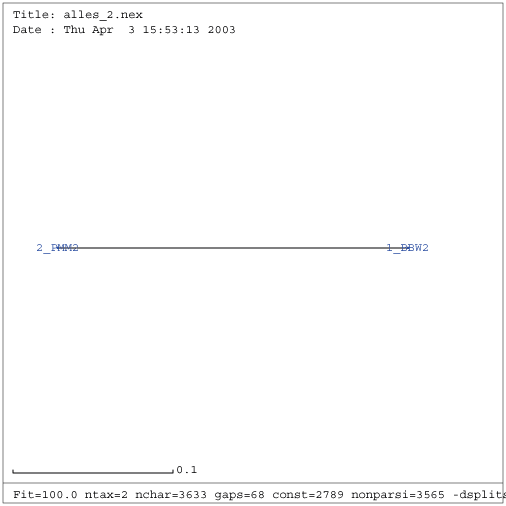
|
| Verwandtschaftsbaum RNA 1 (keine Eliminierung vorgenommen) | Verwandtschaftsbaum RNA 2 (keine Eliminierung vorgenommen) |
Erwartungsgemäß ist eine Eliminierung bei nur zwei gefundenen Sequenzen der Gattung Fabavirus nicht sinnvoll. Ebenso sind konservierte Sekundärstrukturen bzw. kompensatorische Mutationen nicht zu erwarten. Deshalb wird die Gattung der Fabaviren im Folgenden nicht weiter betrachtet und von der Untersuchung ausgeschlossen.
Gattung Nepovirus
| RNA 1 | RNA 2 |
|---|---|
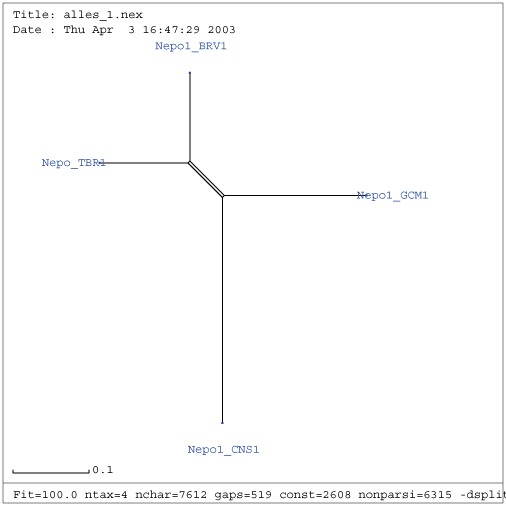
|

|
| Verwandtschaftsbaum RNA 1 nach Entfernung der Viren AF016626, NC_003509, NC_003615, U50869 und NC_003840 | Verwandtschaftsbaum RNA 2 nach Entfernung der Viren AY017339, NC_003502, NC_003623, NC_003839 |
Berechnung der Sekundärstruktur
Mit dem Programm RNAfold als Teil des Vienna RNA Packages kann aus geeigneten Quelldaten die zu erwartende Sekundärstruktur von RNA-Strängen errechnet werden. Das Tool nutzt Sequenzdateien im Vienna-Format die mehrere Sequenzen umfassen, um die Berechnung durchzuführen.
Als Ausgabedateien entstehen Dotplots (PostScript) für jedes RNA-Genom und jeweils ein Minimum-Free-Energy-File welches die energetischen Zustände in der Struktur beschreibt. Der Anteil der Dot-Bracket-Notation innerhalb der Datei beschreibt öffnende und schließende Basenpaare einer Struktur.
Die o.g. Dateien können über die unten stehenden Links heruntergeladen werden.
Gattung Comovirus
| RNA 1 | RNA 2 |
|---|---|
|
Dotplots (gezippte PostScripts) RNAfold-Ausgabedatei |
Dotplots (gezippte PostScripts) RNAfold-Ausgabedatei |
Gattung Nepovirus
| RNA 1 | RNA 2 |
|---|---|
|
Dotplots (gezippte PostScripts) RNAfold-Ausgabedatei |
Dotplots (gezippte PostScripts) RNAfold-Ausgabedatei |
Suche nach konservierten Strukturen
Anschließend wird die Suche nach konservierten Strukturen durchgeführt. Konservierte Sekundärstrukturen werden trotz Mutationen innerhalb der Sequenz erhalten. D.h. wenn Basen mutieren, mutiert die jeweilig komplementäre Base ebenfalls. Somit kommt es wiederum zum Aufbau von Wasserstoffbrückenbindungen zwischen beiden Nucleotiden. Diese Bindung hat zur Folge, dass sich die gleiche Sekundärstruktur entfaltet.
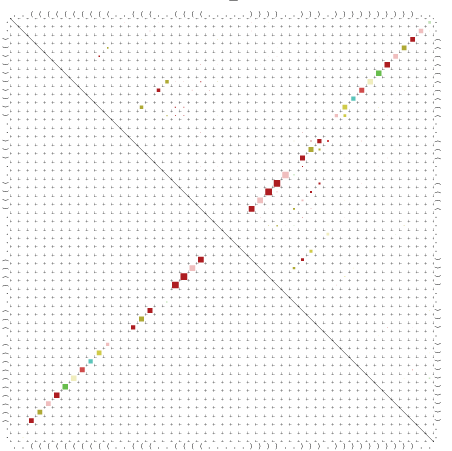
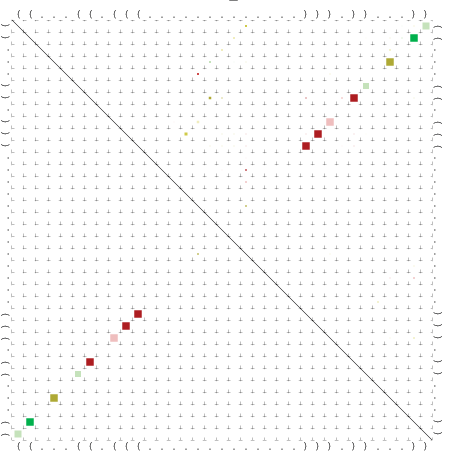
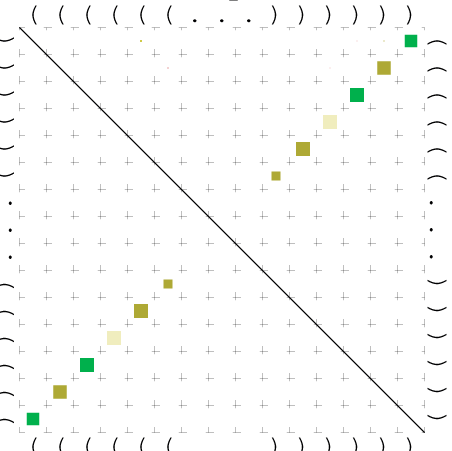
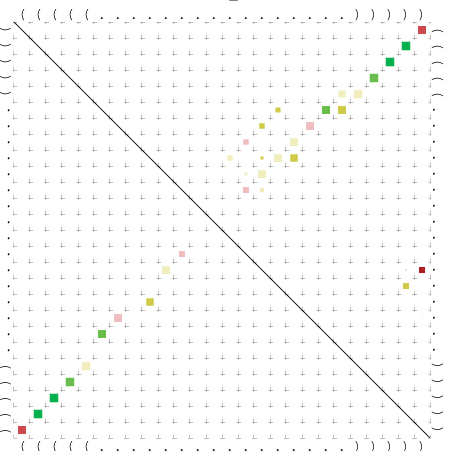
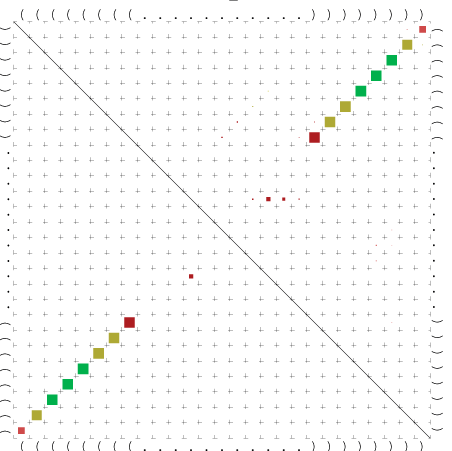
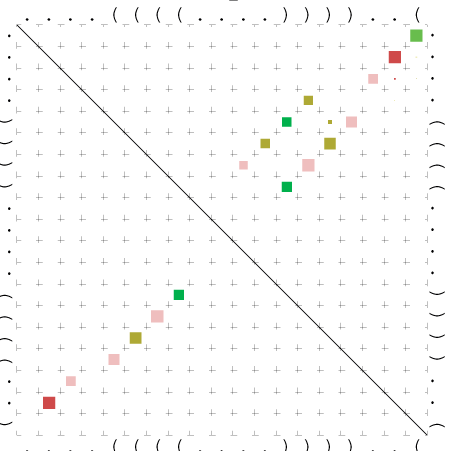
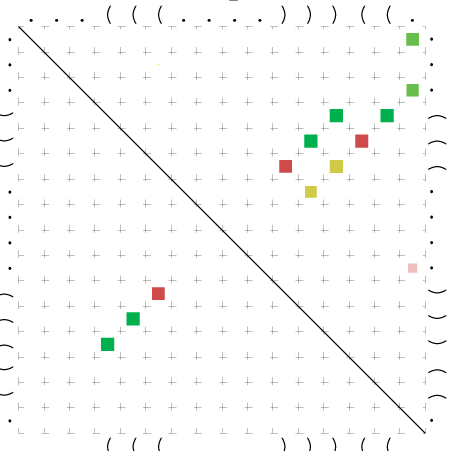
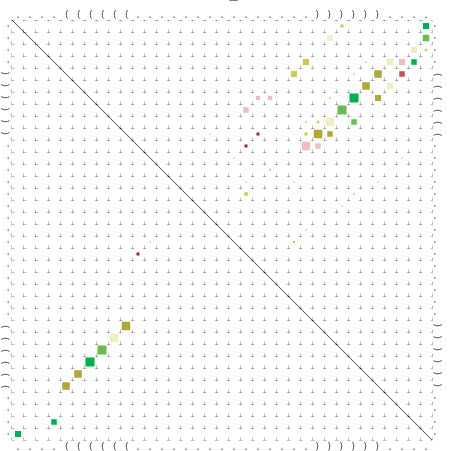
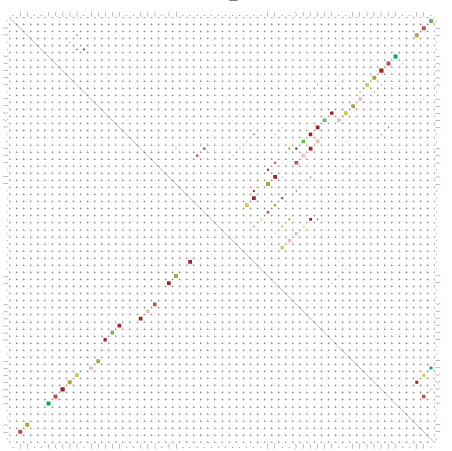
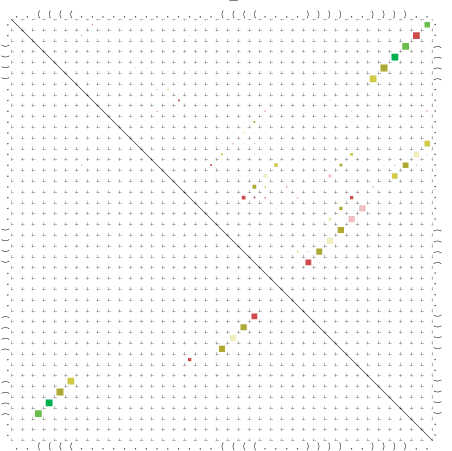
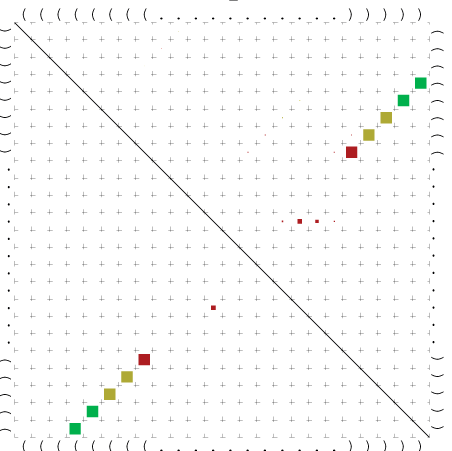
Zur Bewertung ist entscheidend, dass die Konservierung bei möglichst vielen Einzelsträngen (unterschiedlicher Viren) auftritt. Häufiger konservierte Strukturen weisen auf strukturell bedeutsame Teilstränge hin. Bei diesen Abschnitten ist nicht die Codierung der Sequenz entscheidend, sondern die räumliche Struktur, in der die Fragmente falten.Das Tool alidot ermöglicht in Zusammenarbeit mit dem Script anote.pl die Markierung der Sequenzabschnitte, die wahrscheinlich konservierte Sekundärstrukturen enthalten. Hierfür müssen zunächst die Dotplots der einzelnen Sequenzen zu einem gemeinsamen Dotplot verrechnet werden. Anhand des resultierenden Dotplots und der Visualisierungssoftware AliDot werden zu erwartende Sequenzbereiche visuell ausgewählt. Der Dotplot wird folgendermaßen interpretiert.
Die markierten Sequenzabschnitte werden anschließend mit Hilfe des Tools extract.sh aus der Gesamtsequenz ausgeschnitten. Die Fragmente können entsprechend ihrer Mutationswahrscheinlichkeit innerhalb aller Sequenzen, die im Gesamt-Dotplot enthalten sind, markiert werden. Eine Auswahl der besten Ergebnisse sind unten dargestellt. Je dunkler dabei die Färbung der Kreise bzw. Nucleotide ist, desto höher ist die Wahrscheinlichkeit einer konservierten Sekundärstruktur. Zur Bewertung sind die genutzten Regionen des Dotplots ebenfalls abrufbar.
Gattung Comovirus
| RNA 1 | RNA 2 |
|---|---|
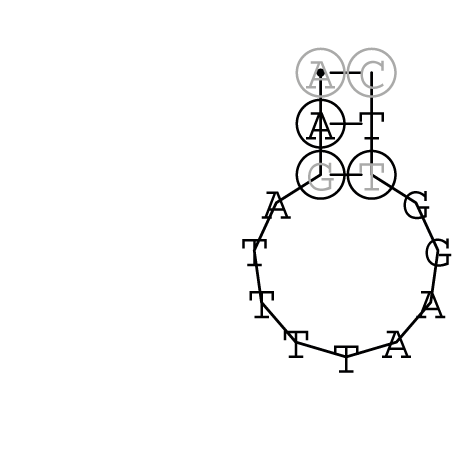 Nucleotid 331 - 345 Dotplot 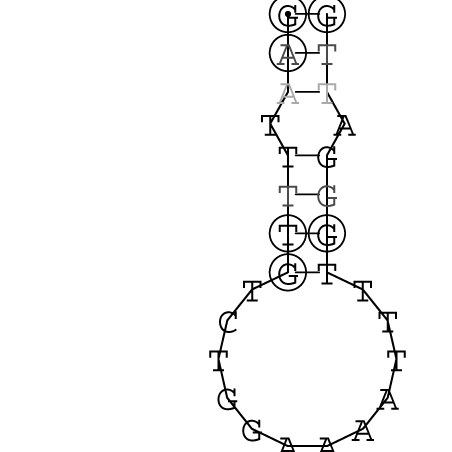 Nucleotid 1552 - 1549 Dotplot 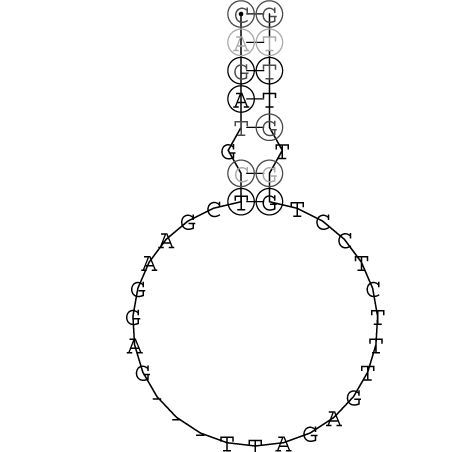 Nucleotid 1718 - 1758 Dotplot 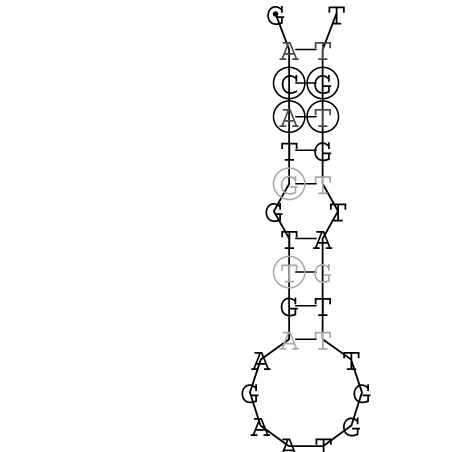 Nucleotid 4192 - 4221 Dotplot 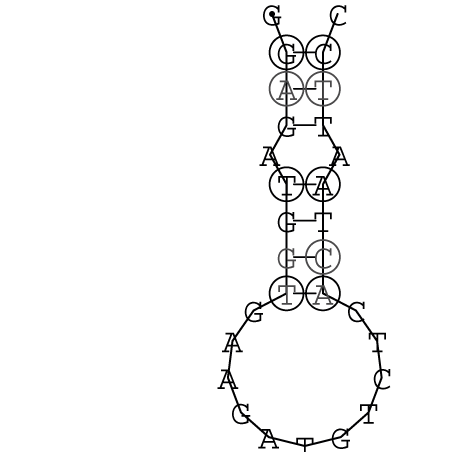 Nucleotid 4384 - 4411 Dotplot 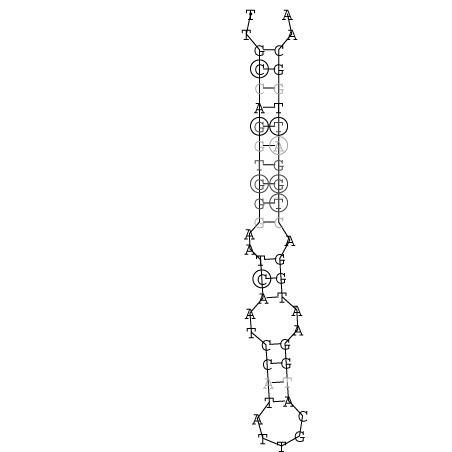 Nucleotid 4646 - 4695 Dotplot |
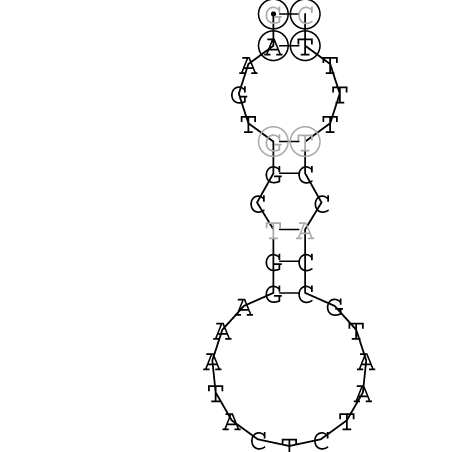 Nucleotid 3456 - 3580 Dotplot |
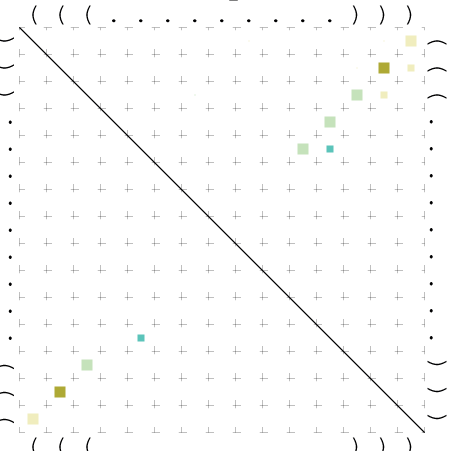{kind=link}
{kind=link}
{kind=link}
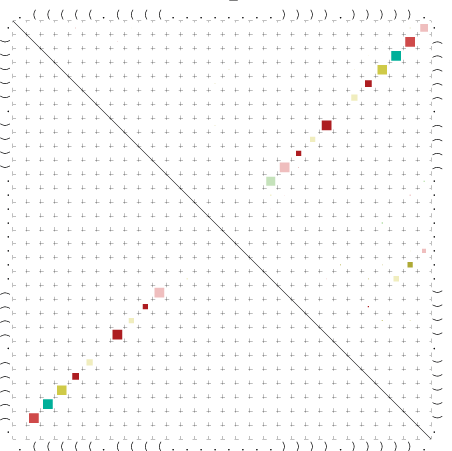{kind=link}
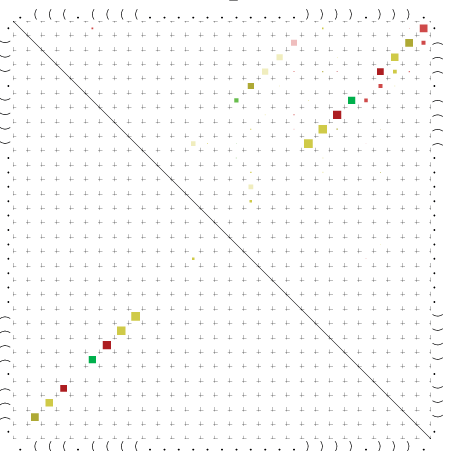{kind=link}
{kind=link}
{kind=link}
Gattung Nepovirus
| RNA 1 | RNA 2 |
|---|---|
 Nucleotid 847 - 868 Dotplot 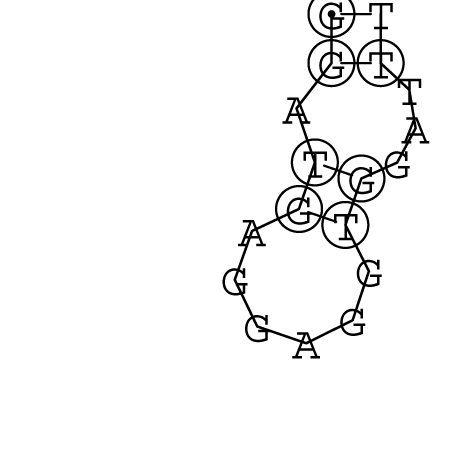 Nucleotid 4404 - 4421 Dotplot 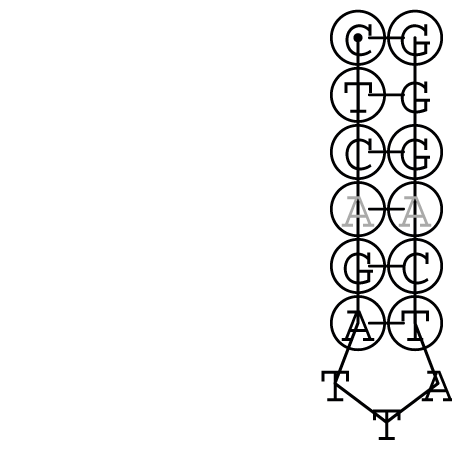 Nucleotid 6212 - 6226 Dotplot 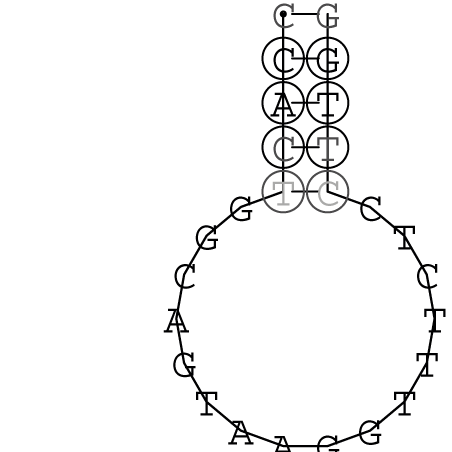 Nucleotid 7229 - 7254 Dotplot 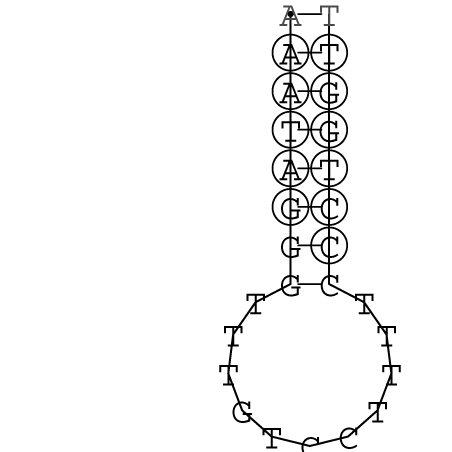 Nucleotid 7516 - 7542 Dotplot |
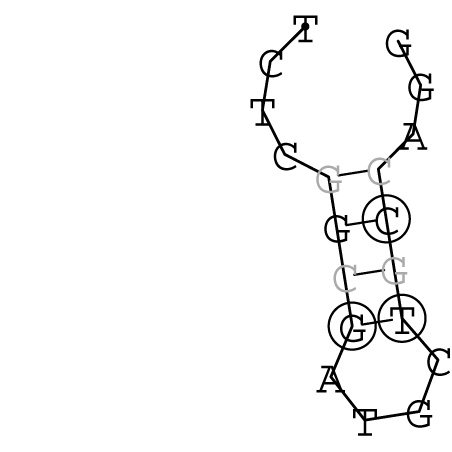 Nucleotid 341 - 359 Dotplot 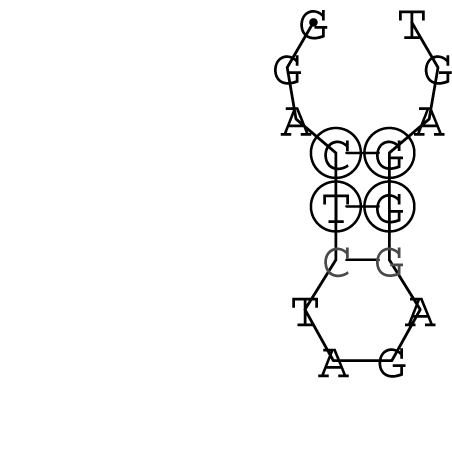 Nucleotid 740 - 755 Dotplot 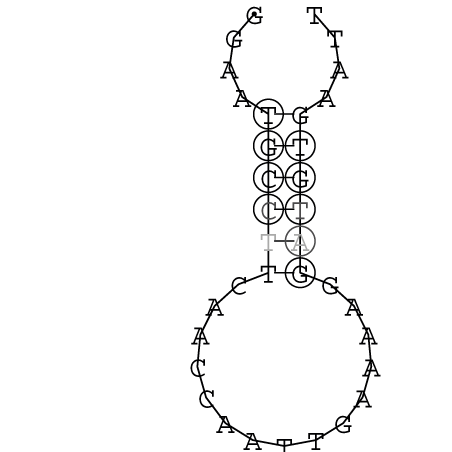 Nucleotid 1257 - 1291 Dotplot 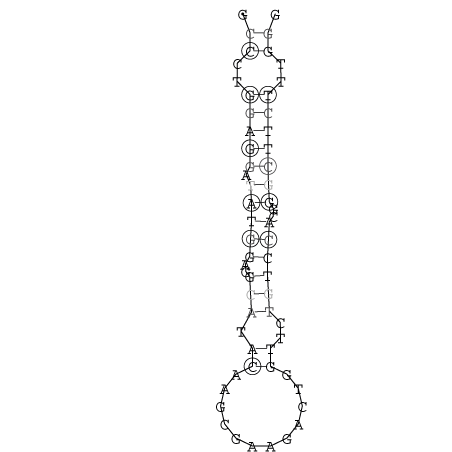 Nucleotid 2328 - 2387 Dotplot 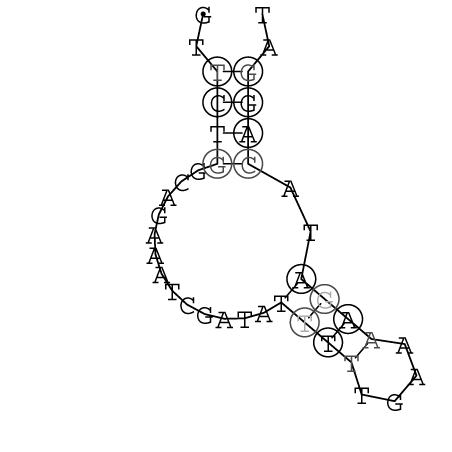 Nucleotid 2916 - 2954 Dotplot 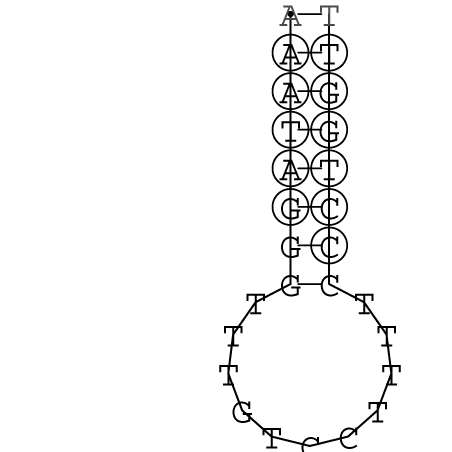 Nucleotid 4793 - 4819 Dotplot |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}