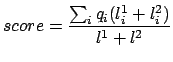 |
||
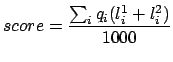 |
||
![\includegraphics[width=1.5\textwidth]{gfx/meme_out.eps}](img12.png) |
| Programme | Input | Output | Anmerkungen | Skript für matCompare | Skripte für Grafiken (*.data) |
| rVista | fasta | keiner | Error-Message erhalten, da Sequenzen zu kurz | leer | leer |
| MEME | fasta | meme | meme2pwm.pl | meme2data_html.pl | |
| AlignACE | fasta | aa | ace2pwm.pl | ace2data.pl | |
| consensus | multifasta | cons | für uns nicht weiter relevant | consensus2pwm.pl | consensus2data.pl |
| bioprospector | fasta | biop | bio2mat.pl | bio2data.pl | |
| YMF | ymfdata2mat.pl | ymf2data.pl;skoda2normal.pl | |||
| nestedMICA | email geschickt | leer | leer | ||
| TFSearch | leer | tfsearch2data.pl | |||
| pwmatch | transfacpwmatch.pl | transfacpwmatch_data.pl |
![\includegraphics[scale=0.5]{gfx/uebersicht.eps}](img2.png)
![\includegraphics[scale=0.5]{gfx/abb1.ps}](img5.png)
![\includegraphics[scale=0.5]{gfx/abb2.eps}](img13.png)