Übersicht der Arbeitsschritte zur Untersuchung der Sequenz T0134
Schritt 1) Alignmentsuche in Datenbank
Schritt 2) Fold-Recognation
Schritt 3) PDB-Datenbank
Schritt 4) FUGUE Alignment
Schritt 5) Modeller erzeugt 3D-Model
Schritt 6) Ermittlung der Aussagekraft des erstellten Modells
Schritt 7) Energievergleich mit Originalprotein
Schritt 8) Vergleich der Fold-Recognation-Tools
Sequenzdaten
T0134 Delta-adaptin appendage domain, human (251 letters)
GEPVQNGAPEEEQLPPESSYSLLAENSYVKMTCDIRGSLQEDSQVTVAIVLENRSSSILK GMELSVLDSLNARMARPQGSSVHDGVPVPFQLPPGVSNEAQYVFTIQSIVMAQKLKGTLS FIAKNDEGATHEKLDFRLHFSCSSYLITTPCYSDAFAKLLESGDLSMSSIKVDGIRMSFQ NLLAKICFHHHFSVVERVDSCASMYSRSIQGHHVCLLVKKGENSVSVDGKCSDSTLLSNL LEEMKATLAKC
Schritt 1) Alignmentsuche in einer Proteindatenbank
Die Alignmentsuche mit dem Programm BlastP (NCBI) erbrachte ein Alignment mit
einem Score von 29 (E-value=2.2). Dieser Score ist zu gering, um sich darauf
verlassen zu koennen. Das Ergebnis wird daher nicht weiter beachtet. (Link)
zur Übersicht
Schritt 2) Fold-Recognation
Im diesem Schritt wird erneut nach einem homologen Protein gesucht. Die Auswertung
des Tools FUGUE ergibt gleich zwei moegliche homologe Proteine fuer unsere
Sequenz. Ihr PDB-Code ist 1qts und 1e42.
zur Übersicht
Schritt 3) PDB-Datenbank
Anhand des PDB-Codes kann fuer diese beiden Proteine jeweils das pdb-File heruntergeladen werden, welches das 3D-Modell dieser beiden homologen Proteine enthaelt. Diese Files werden im Folgenden benoetigt, um aus unserer Sequenz ein dreidimensionales Modell erstellen zu koennen.
Struktur-Modell von 1qts
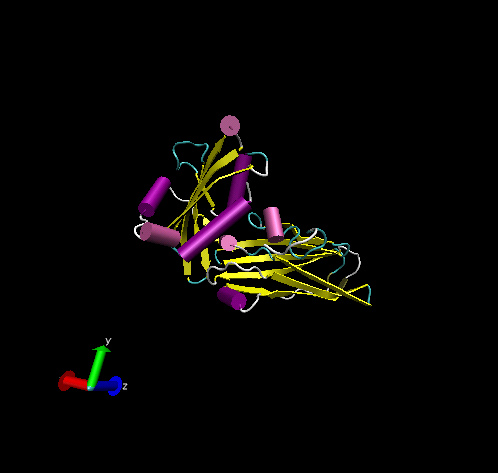
Struktur-Modell von 1e42
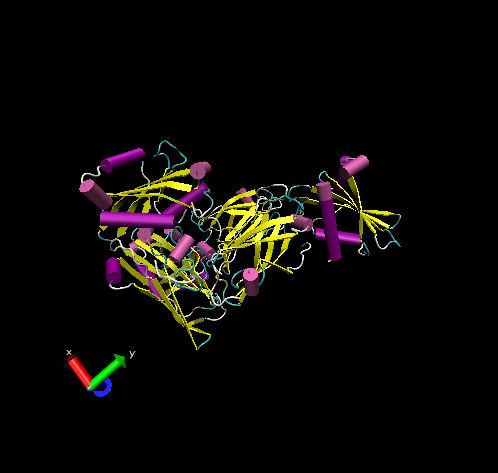
zur Übersicht
Schritt 4) FUGUE Alignment
Auf der FUGUE-Seite wird nun ein Alignmnet-Tool verwendet, welches unsere
Ausgangssequenz mit den beiden 3D-Strukturfiles der homologen Proteine alignt.
Dabei entsteht ein .pir-File (align_1qts_T0134.pir), welches ebenfalls der
3D-Modellerzeugung dient. Der Inhalt des .pir-Files ist im Folgenden dargestellt:
zur Übersicht
>P1;1qts
structureX:1qts
GSPGIRLGSSEDNFARFVCKNNGVLFENQL--LQIGLKSEFRQ-NLGRMFIFYGNKTSTQFLNFTPTLIC
ADDLQTNLNLQTK-------PVDPTVDGG--AQVQQVVNIECI---SDFTEAPVLNIQFRYGGTFQNVSV
KLPITLNKFFQPTEMASQDFFQRWKQLSNPQQEVQNIFKAKHPMDTEITKAKIIGFGS-ALLEEVDPNPA
NFVGAGIIHTKTTQIGCLLRLEPNLQAQMYRLTLRTSKDTVSQRLCELLSEQF---*
>P1;4542
Sequence: 4542
GEPVQNGAPEEEQLPP--ESSYSLLAENSYVKMTCDIRGSLQEDSQVTVAIVLENRSSSILKGMELSVL-
-DSLNARMARPQGSSVHDGVPVPFQLPPGVSNEAQYVFTIQSIVMAQKLKGTLSFIAKNDEGATHEKLDF
RLHFSCSSYLITTPCYSDAFAKL---LESGDLSMSSIKVDGIRMSFQNLLAKICFHHHFSVVERVD----
---SCASMYSRSIQ-GHHVCLLVKKGENSVSVDGKCSDSTLLSNLLEEMKATLAKC*
>P1;1e42
structureX:1e42: 705 :A: 937 :A: : : :
GYV---APKAVWLPAVKAKGLEISGTFTHR----------QGHIYMEMNFTNKALQHMTDFAIQFNKNS
-------FGVIPSTPLAIHTPLMPNQSIDVSLPLNTLGPVMKMEPLNNLQVAVKNNIDVFYFSCLIPLNV
LFVE---DGKMERQVFLATWKDIPNENELQFQIKECHLNADTVSSKLQNNNVYTIAKRNVEGQDMLYQSL
KLTNGIWILAELRIQPGNPNYTLSLKCRAPEVSQYIYQVYDSILKN*
>P1;T0134
sequence
EPVQNGAPEEEQLPPESSYSLLAENSYVKMTCDIRGSLQEDSQVTVAIVLENRSSSILKGMELSVLDSL
NARMARPQGSSVHDGVPVPFQLPPGVSNEAQYVFTIQSIVMAQKLKGTLSFIAKNDEGATHEKLDFRLHF
SCSSYLITTPCYSDAFAKLLES-GDLSMSSIKVDGIRMSFQNLLAKICFHHHFSVVERVDSCASMYSRSI
QGHH-----VCLLVKKGENSVSVDGKCSDSTLLSNLLEEMKATLAK*
Schritt 5) Modeller erzeugt 3D-Model
Im Modeller wird nun anhand des soeben erzeugten .pir-Files und der .pdb-Struktur
jeweils eines der beiden homologen Proteine ein dreidimensionales Modell
unserer Ausgangsstruktur erzeugt.
Dieses Modell ist mit dem Tool vmd zu betrachten.
Erstelltes Modell mit 1qts
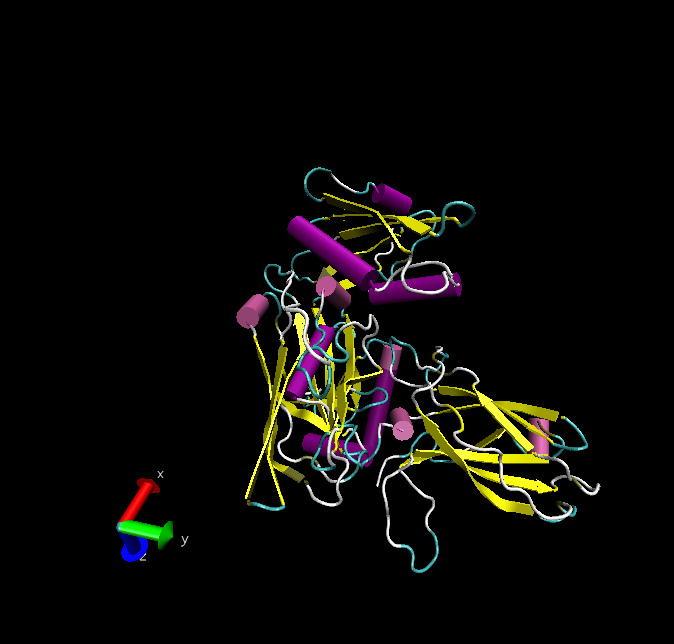
Erstelltes Modell mit 1e42
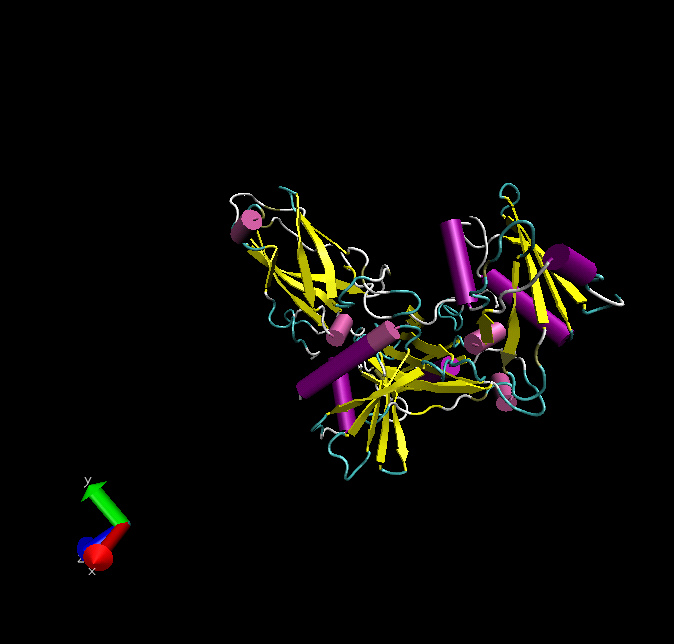
zur Übersicht
Schritt 6) Ermittlung der Aussagekraft des erstellten Modells
Mit Hilfe des Tools "profit" wird aus dem erstellten Modell, sowie aus dem
jeweiligen 3D-Modell der homologen Proteine der RMS-Wert berechnet. Das ermoeglicht
uns zwischen den beiden anfangs ermittelten homologen Proteinen und den daraus
erstellten 3D-Modellen unserer Sequenz das aussagekräftigere Modell ausfindig zu machen.
zur Übersicht
Ergebnis:
Vergleich der modeller.pdb mit naehester_struktur.pdb [profit]
1qts: RMS = 11.74 > 10 Modell nicht verwendbar da schlechte Vorhersage
1e42: RMS = 10.17 = 10 Modell verwendbar (Zone fuer t0134 1-233)
Das aus dem Protein mit dem PDB-Code 1e42 gewonnene 3D-Modell ist mit einem RMS-Wert von fast 10 ein brauchbares Modell und wird deshalb als Ergebnis des Auswertungsprozesses angesehen.
Schritt 7) Energievergleich mit Originalprotein
Das Programm Prosa erstellt eine Ansicht der innermolekularen Kräfte
eines Proteins. So kann das erzeugte Model ein weiteres Mal auf seine Aussagekraft
hin untersucht werden. Im Folgenden dargestellt ist das Prosa-Ergebnis, in dem die
untere energieärmere Kurve unser Model, die obere Kurve das homologe Protein darstellt.
Aufgrundder RMS-Werte haben wir diesen Schritt nur für das aussagekräftige Protein 1e42
durchgeführt.
Erstelltes Modell mit 1e42
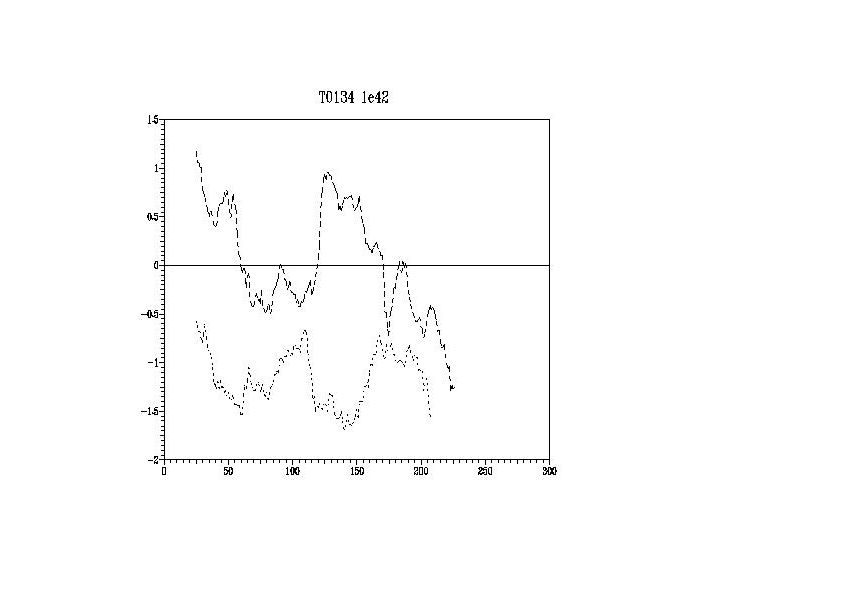
Es ist festzustellen, dass das Energiediagramm unserem erstellten 3D_Modell keine
guten Eigenschaften zuweist. Die Kurve liegt beträchtlich im negativen Bereich,
allerdings ist das auch teilweise bei unserem homologen Ausgangsprotin der Fall,
weshalb das nicht überzubewerten ist.
zur Übersicht
Schritt 8) Vergleich der Fold-Recognation-Tools
Neben FUGUE existieren noch weitere Tools, die es ermoeglichen eine Fold-Recognation durchzufuehren. Wir haben uns entschieden fuer dieses Beispiel die moeglichen Ergebnisse dieser anderen Tools zu Vergleichszwecken zu ermitteln und die naechsten Sequenzen T0142-T0166 mit dem Tool weiter zu bearbeiten, welches uns hier das beste Ergebnis liefert.
Tool 2) 123D_foldrec.
bester Kandidat mit Z-score=5.96: 1e0ta
weiterer Weg wie oben,
Vergleich der modeller.pdb mit 1e0ta.pdb [profit]
-> RMS = 14.829 > 10 kein besseres Modell gefunden
-> zur Auswertung
Tool 3) 3dpssm_foldrec
bester kandidat ist 1b9k mit e-value: 0,173
schlechterer RMS-Wert 18,706 als bei foldrec durch fugue
ergo bleiben wir bei unserer ergebnislage
-> zur Auswertung
Tool 4) Swiss modeller
Ergebnis: "The degree of similarity of your sequence with proteins of
known 3D structure may be to low."
Das beste Ergebnis mit einem RMS-Wert von 10,17 lieferte uns FUGUE. Dieses Tool
wird uns also weiter begleiten. Als Fazit lässt sich festhalten, dass wir ein
einigermassen annehmbares 3D-Modell für unsere Sequenz auf diesem Weg erzeugen konnten.
Das gibt Hoffnung für die Zukunft.
zur Übersicht