Kurze, circa 70 bis 120 Nukleotide lange, nicht codierende RNA, die aufgrund ihrer Groesse microRNA genannt werden, spielen auf der post-transkriptionalen Ebene eine wichtige Rolle beim Gensilencing. Diese microRNAs oder auch miRNAs kommen in Pflanzen, Tieren und in einigen Viren vor und sind aufgrund ihrer hohen Konserviertheit fuer phylogenetische Analysen geeignet. Phylogenetische Analysen werden zur Klaerung evolutionaerer Verwandschaftsverhaeltnisse von Organismen verwendet. Wir untersuchen orthologe und paraloge Gene. Orthologe entstehen durch Speziationsereignisse und Paraloge durch Duplikationsereignisse innerhalb einer Spezies. Das bedeutet, dass Orthologe in unterschiedlichen Organismen zu finden sind und Paraloge kommen in der gleichen Spezies vor.
Die im Genom enthaltenen Gene fuer die miRNAs werden von einer RNA-Polymerase II oder III transkribiert und es entsteht ein Primaertranskript mit einen Poly-A-Schwanz am 3'-Ende und einer 5'-Cap-Struktur zur Stabilisierung. Diese pri-miRNA wird von einem Multiprozessor-Komplex , der aus der Rnase III (Drosha) und der dsRNA-Binderprotein DGCR8 geformt wird, im Zellkern prozessiert. Diedaraus entstehende Vorlaeufer-Haarnadelstruktur (precursor hairpin), auch als pre-miRNA abgekuerzt, wird aus dem Zellkern durch Exprotin-5-Ran-GTP ins Cytoplasma ausgeschleust. Im Cytoplasma wird die pre-miRNA Haarnadel durch den RNase Dicer in ihre reife Laenge geschnitten. Dieses Enzym bildet einen Komplex mit dem doppelstraengige RNA bindenden Protein TRBP, wodurch die miRNA-Duplex entwunden und einzelstraengig wird. Der funktionale Strang dieser reifen miRNA wird mit einem RNA-Bindeprotein der Argonaut-Familie beladen und bildet ein RNA-induzierden Silencing-Komplex (RISC), der Ziel-mRNAs durch Spaltung, translationaler Repression oder Deadenylation hemmt.

Im Folgenden werden zwei Moeglichkeiten zur Orthologievorhersage der miRNA-Familien von 15 verschiedenen Insekten betrachtet. Die Unterschiede der manuell gefundenen bzw. der durch das Programm Proteinortho gefundenen Zuordnung der orthologen Sequenzen.
Zunächst wurde in der miRBase-Dantenbank nach den miRNAs folgender Organismen gesucht:
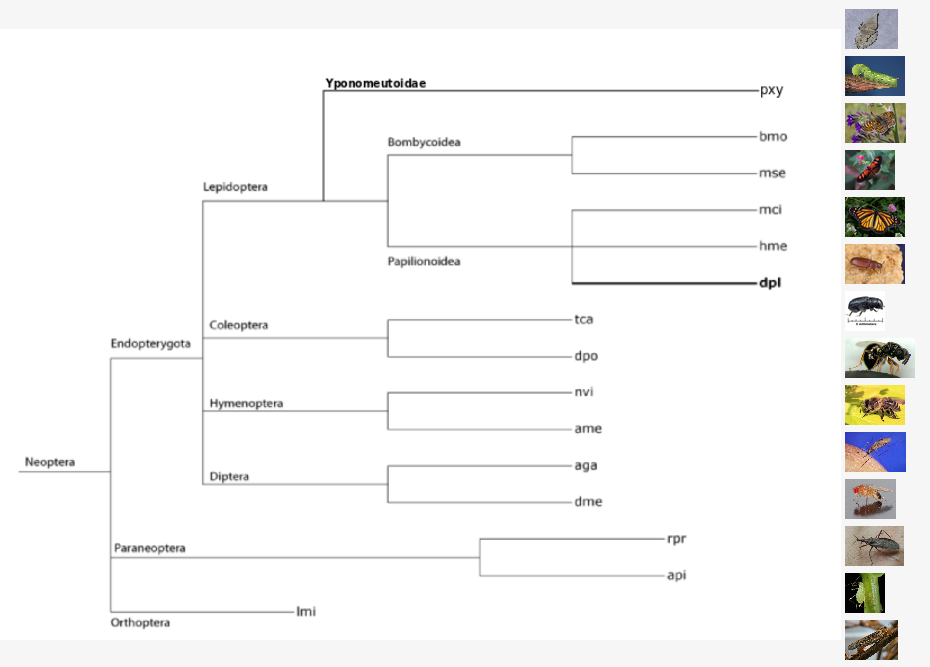
Die pre-miRNAs dieser Organismen wurden, sofern vorhanden, heruntergeladen und gegen die Genome dieser 14 Organismen geblastet. Die gewünschten Daten wurden anschließend extrahiert, in eine bed-Datei überführt, sortiert und gemerged, um überlappende und doppelte Hits herauszufiltern. Aus diesen Dateien wurden anschließend die orthologen und paralogen miRNAs nach folgenden Kriterien ausgewählt: Die Hitlänge sollte mehr als 70 % der Querylänge entsprechen, der Score (Identity * Blasthitlänge) sollte über 6000 sein und der Hit ungefähr die reife miRNA abdeckt.
Zu den ausgewählten paralogen und orthologen miRNAs wurden die MIPF-Familien herausgesucht und anschließend nach diesen sortiert.
Fuer Proteinortho wurden Sequenzen von unseren Organismen aus der miRBase und unsere gefundenen Orthologen und Paralogen in eine Fasta-Datei je Spezies zusammengefuegt. Das Orthologie Erkennungstool Proteinortho findet orthologe Gene in verschiedenen Spezies, indem es Aehnlichkeiten der Sequenzen und Cluster vergleicht, um signifikante Gruppen zu finden. Der Vorteil von diesem Tool ist, dass viele Daten verschiedener Spezies gleichzeitig bearbeitet werden koennen. Um die Genauigkeit der Vorhersage zu erhoehen wird der Befehl mit -synteny ausgefuehrt.
Es wurde ein Skript geschrieben, welches die manuell erstellten Dateien mit den Sequenzen der MIPF-Familien durchgeht und mit den in Proteinortho vorhandenen Sequenzen vergleicht. Im Output werden die Sequenzen innerhalb der Familien aufgefuehrt.