Protokoll Heatmaps & Loss pattern
- Einleitung
- Heatmaps
- Loss pattern
Einleitung
Ziel des Praktikums
Ziel dieses Praktikums war es, anhand der Analyse möglicher Faktoren des Migrationsverhaltens des Monarchfalters Danaus plexippus bioinformatische Arbeitsweisen kennenzulernen und anzuwenden. Dazu wurden Proteine und miRNA des Falters mit den Daten anderer Organismen verglichen.
Die Teilaufgabe unserer Gruppe war es, neu gefundene Paraloge und Orthologe der Organismen mittels Heatmaps darzustellen und zu analysieren und Muster des Verlustes einzelner miRNA-Familien in sogenannten Loss patterns auf Basis der phylogenetischen Beziehungen der Organismen untereinander zu detektieren.
Gruppenmitglieder
- Felix Raith
- Thomas Müller
- Carolin Schimmelpfennig
Rechner
- PRAK03
- PRAK06
Heatmaps
Heatmaps werden dazu verwendet, Werte zweidimensionaler Datensätze mit Farben darzustellen. Historisch wurden Heatmaps schon 1873 von Toussaint Loua genutzt, um statistische Daten der Pariser Stadtteile darzustellen (Quelle: Wikipedia). Die Bezeichnung Heatmap stammt ursprünglich von der intuitiven Assoziation von Farben mit Temperaturen. Heutzutage wird diese Darstellung divers zum Beispiel als Wärmebild, Tree Map oder geclusterte Heatmap verwendet.
In der Bioinformatik finden vor allem die geclusterten Heatmaps Verwendung. Hierbei korrelieren die Werte des Datensatzes mit bestimmten Farben. Mit Hilfe von distanzbasierten Baumdiagrammen (Dendrogrammen) können dann Cluster, also Gruppen, die nach spezifischen Charakteristika aufgeteilt wurden, erkannt werden.
Durch die Farbkodierung in den Heatmaps sind markante Werte großer Datenmengen schnell zu identifizieren.
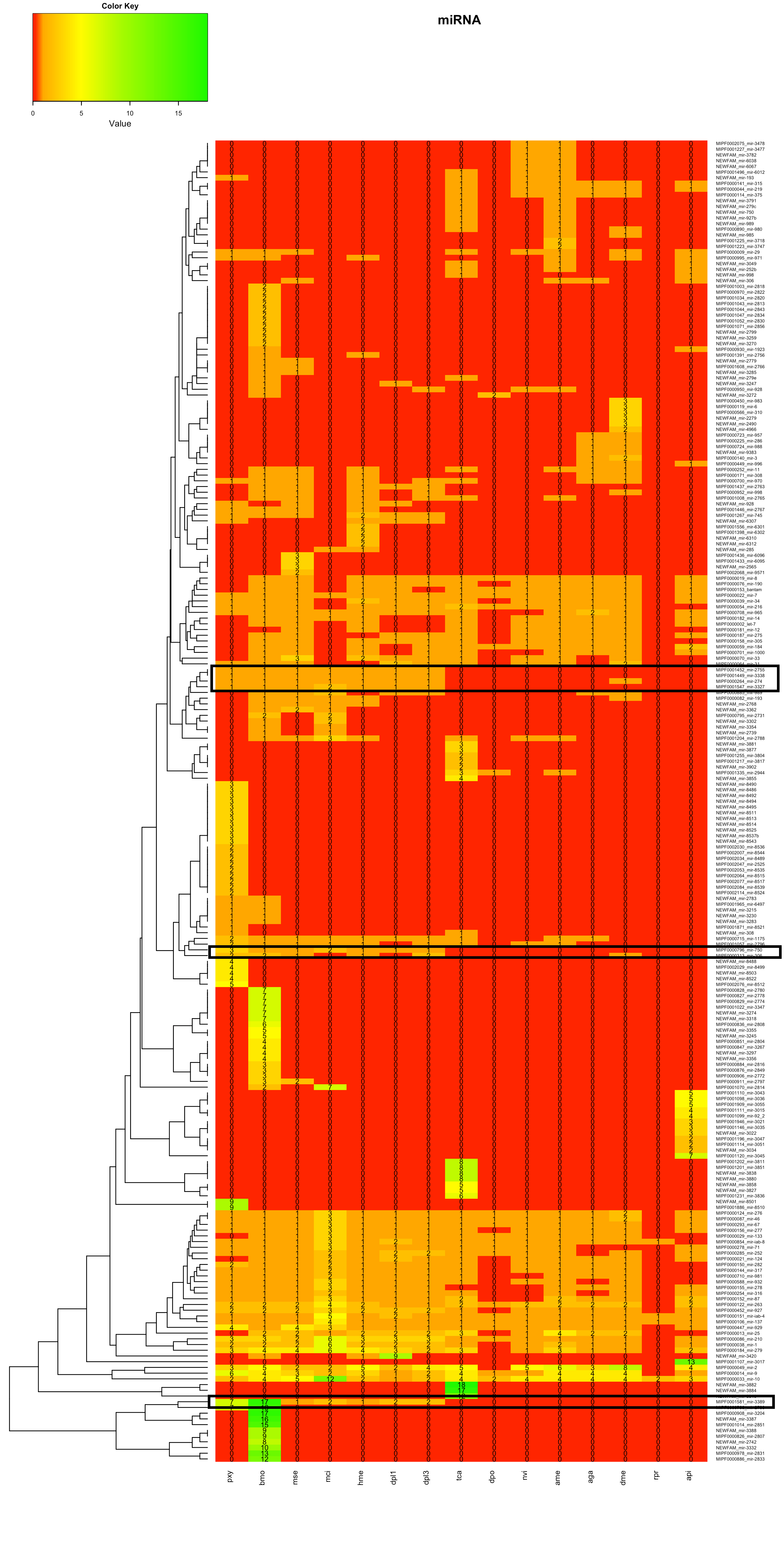
Heatmaps ohne Dendrogramm der Spezies
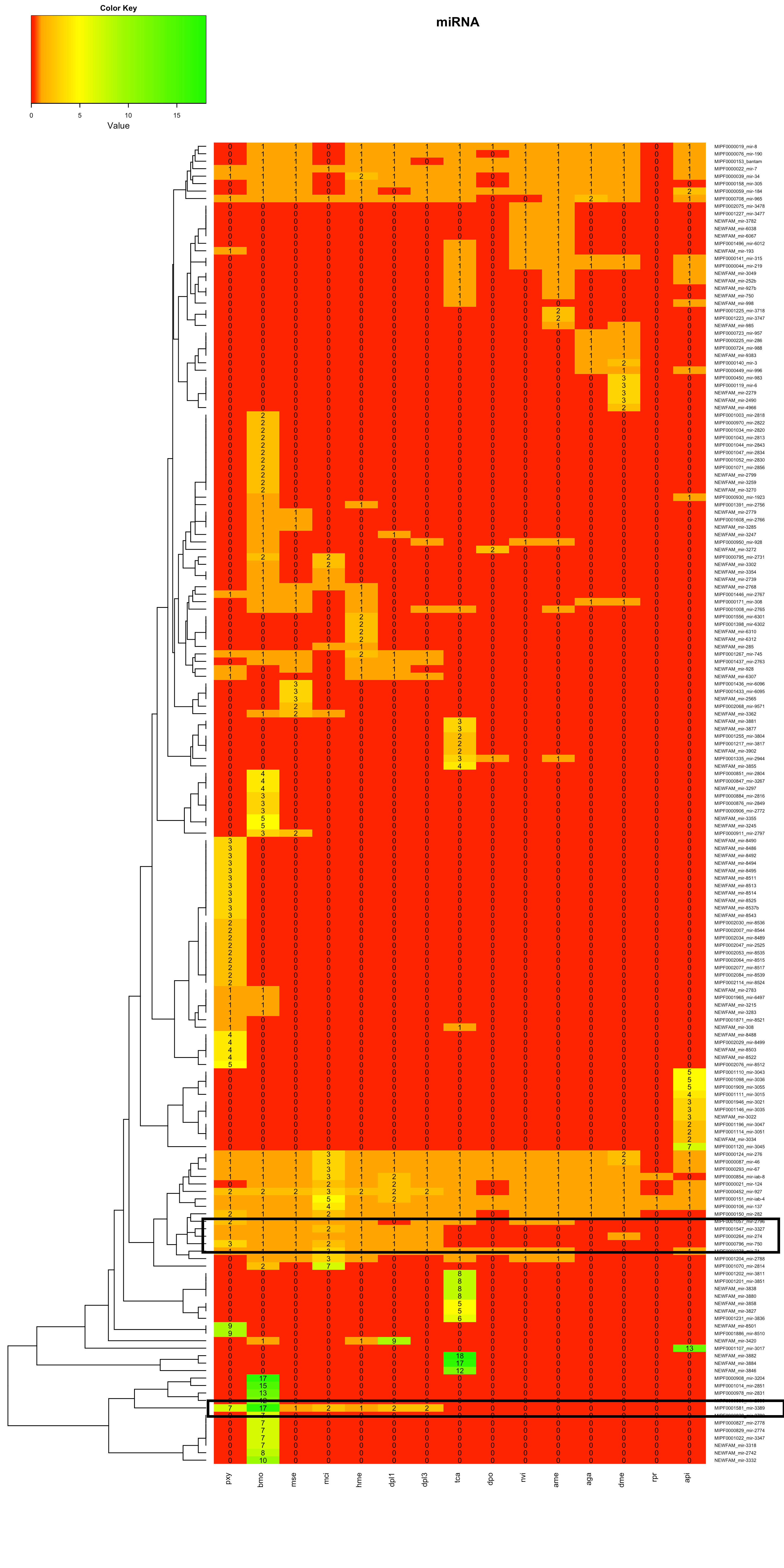
Heatmaps mit Dendrogramm der Spezies
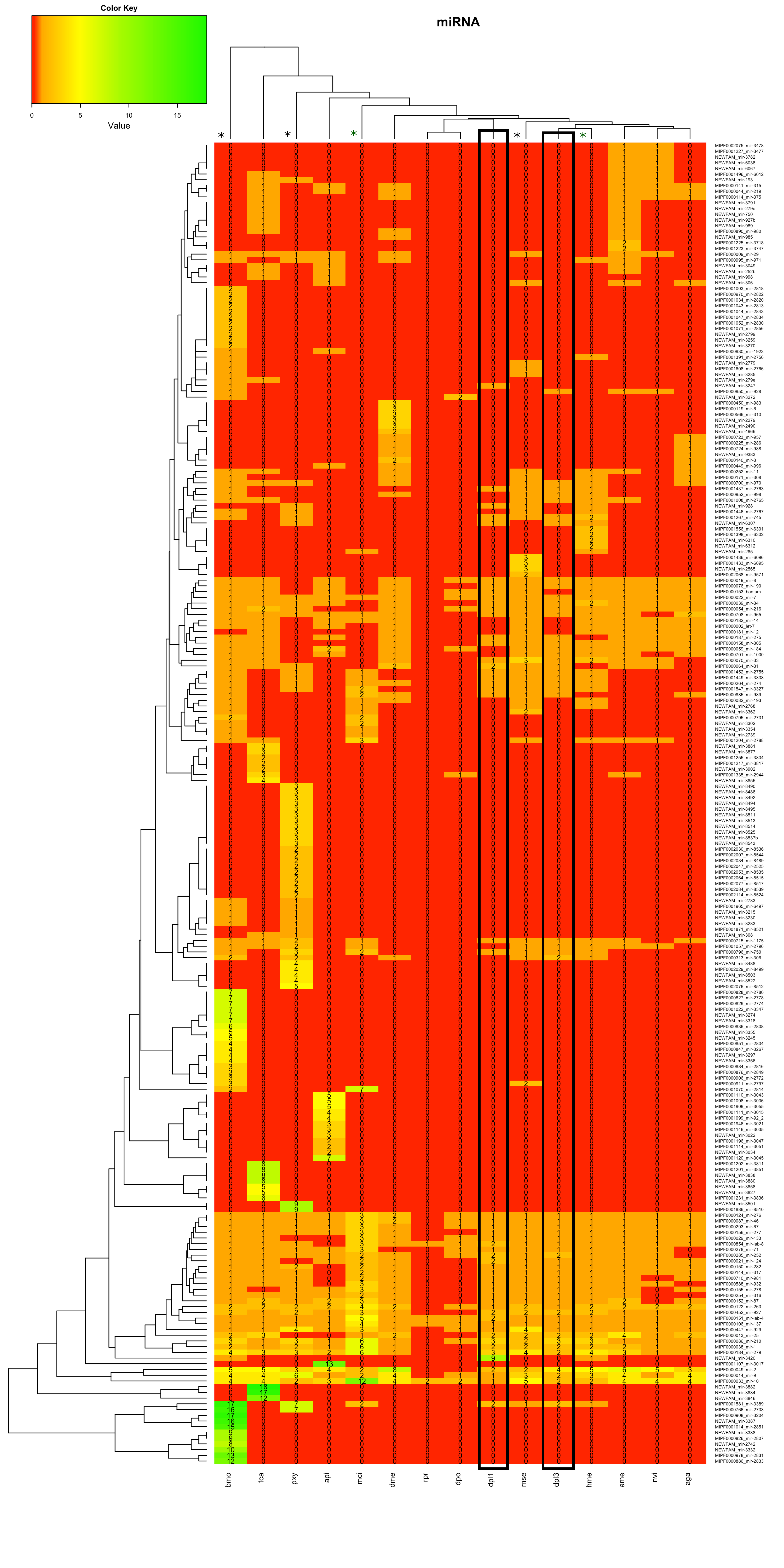
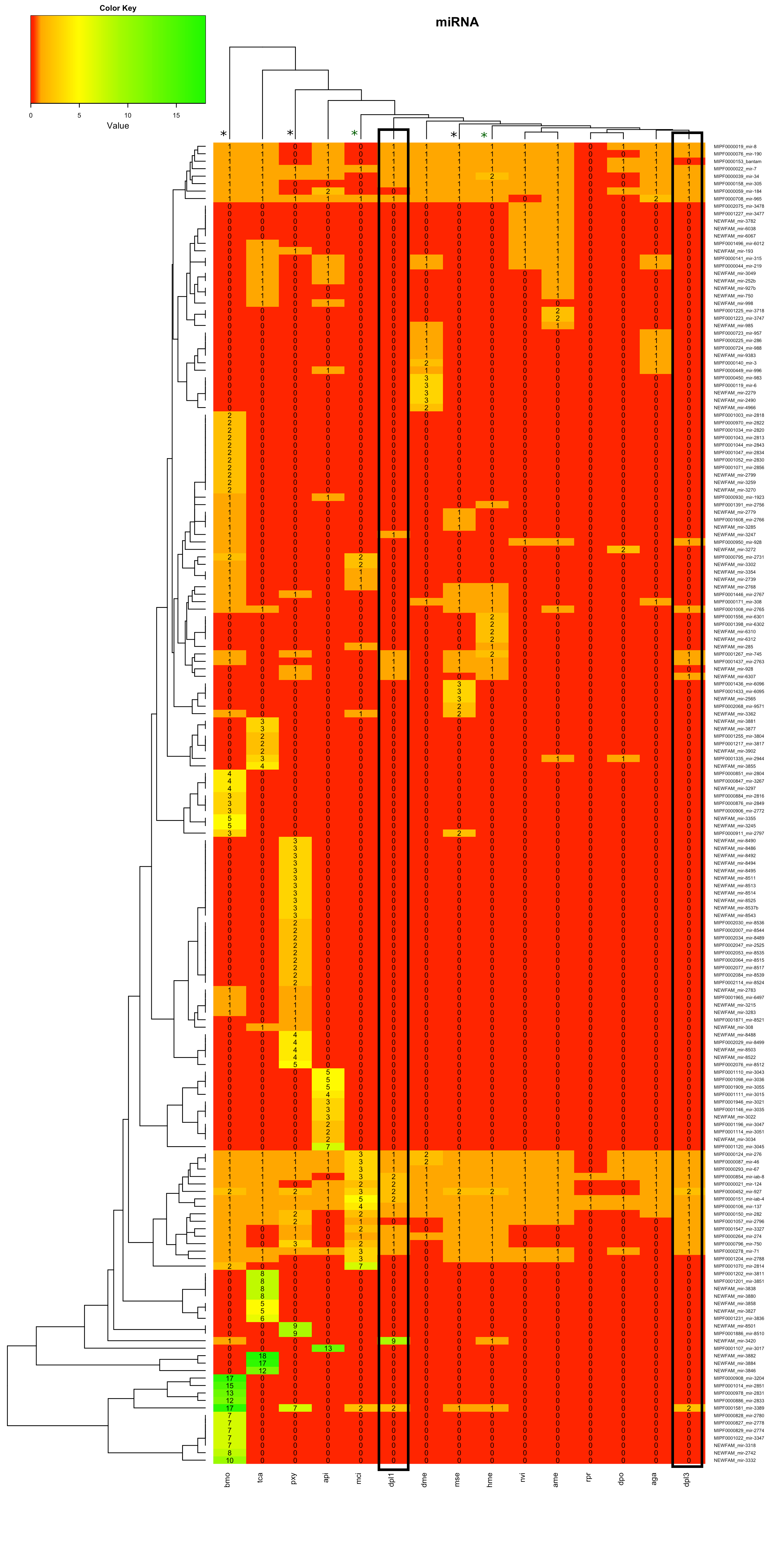
Analyse Heatmaps
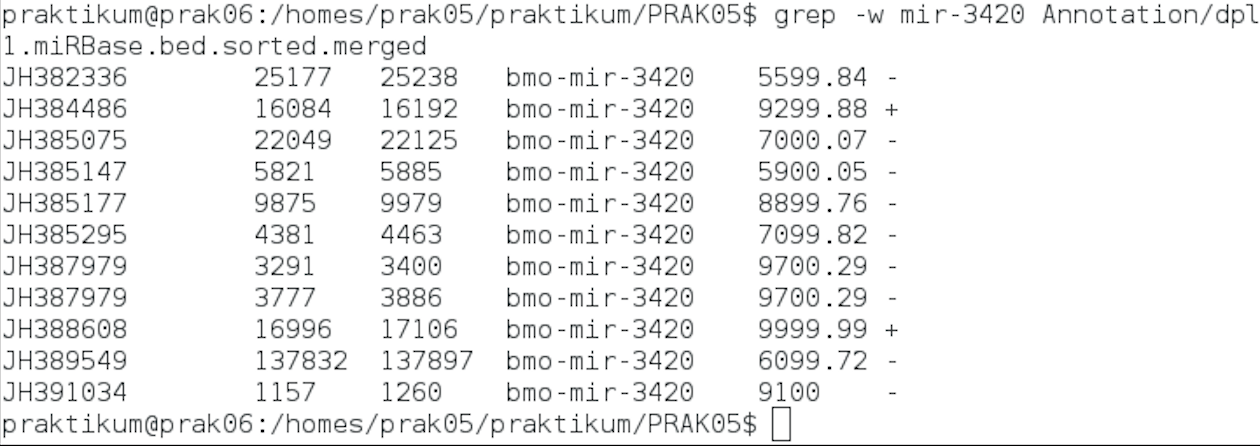
NEWFAM mir-3420
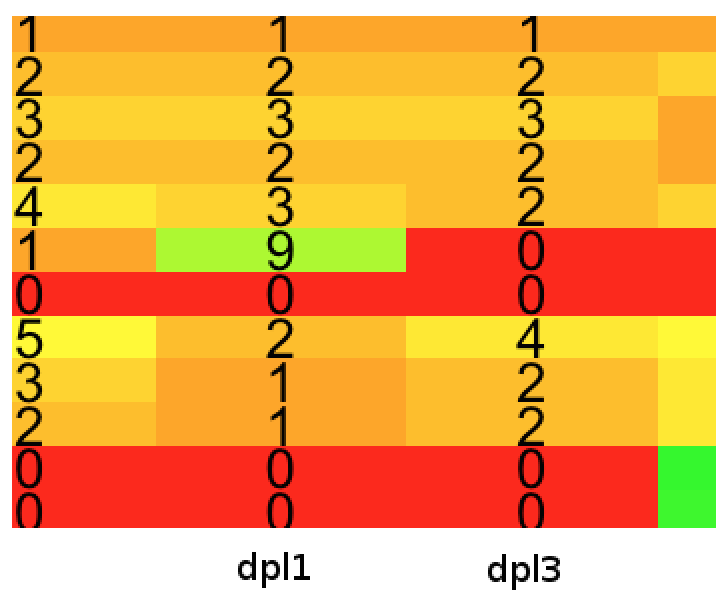

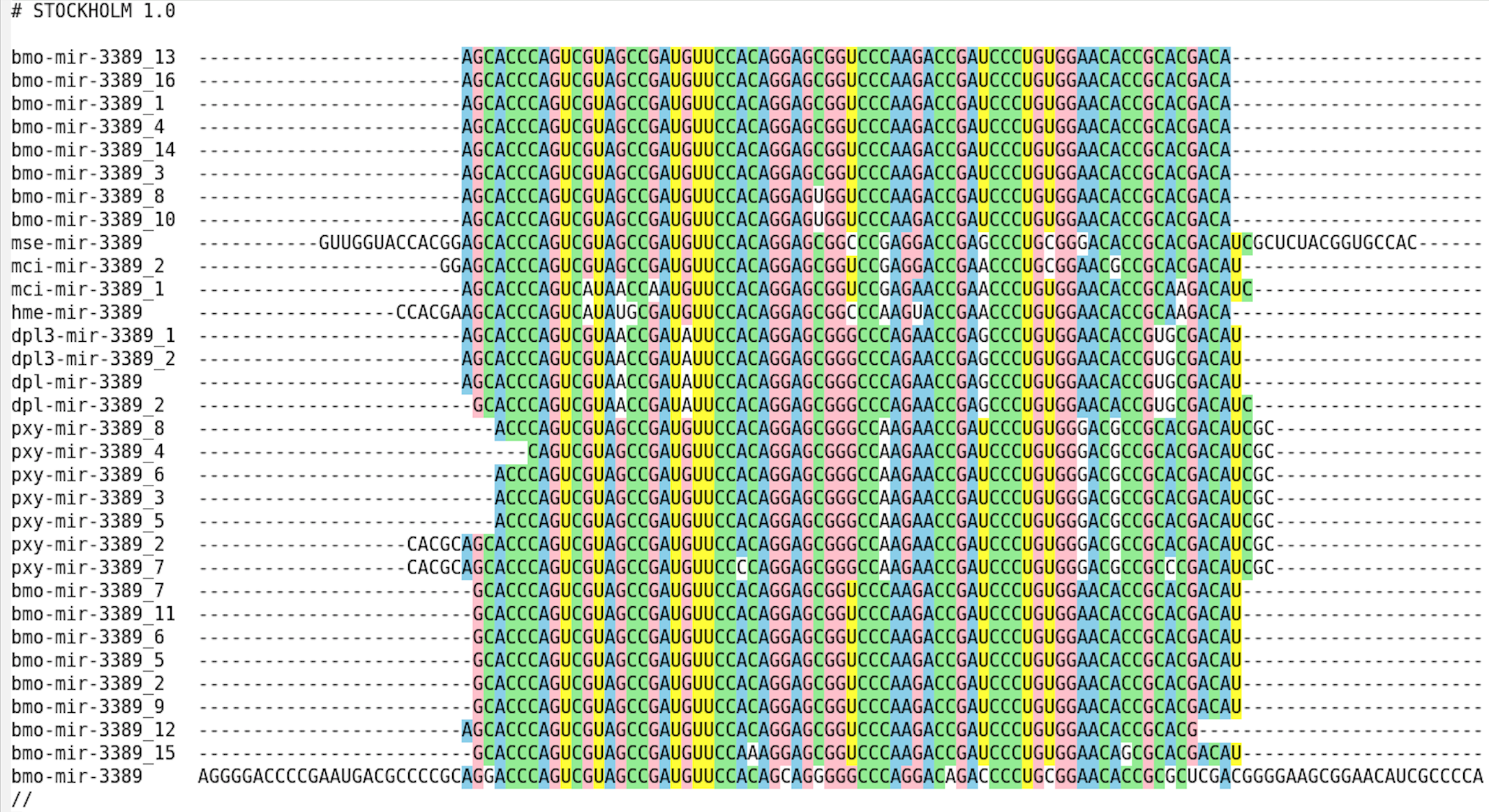
MIPF0001581 mir-3389
In der Heatmap 1.2 war zudem im unteren Teil mehrere Familien des Bombyx mori aufgrund hoher miRNA-Anzahlen auffällig. Zur Bestimmung möglicher Gründe wurde Beispielhaft die MIPF0001581 mir-3389 ausgewählt.
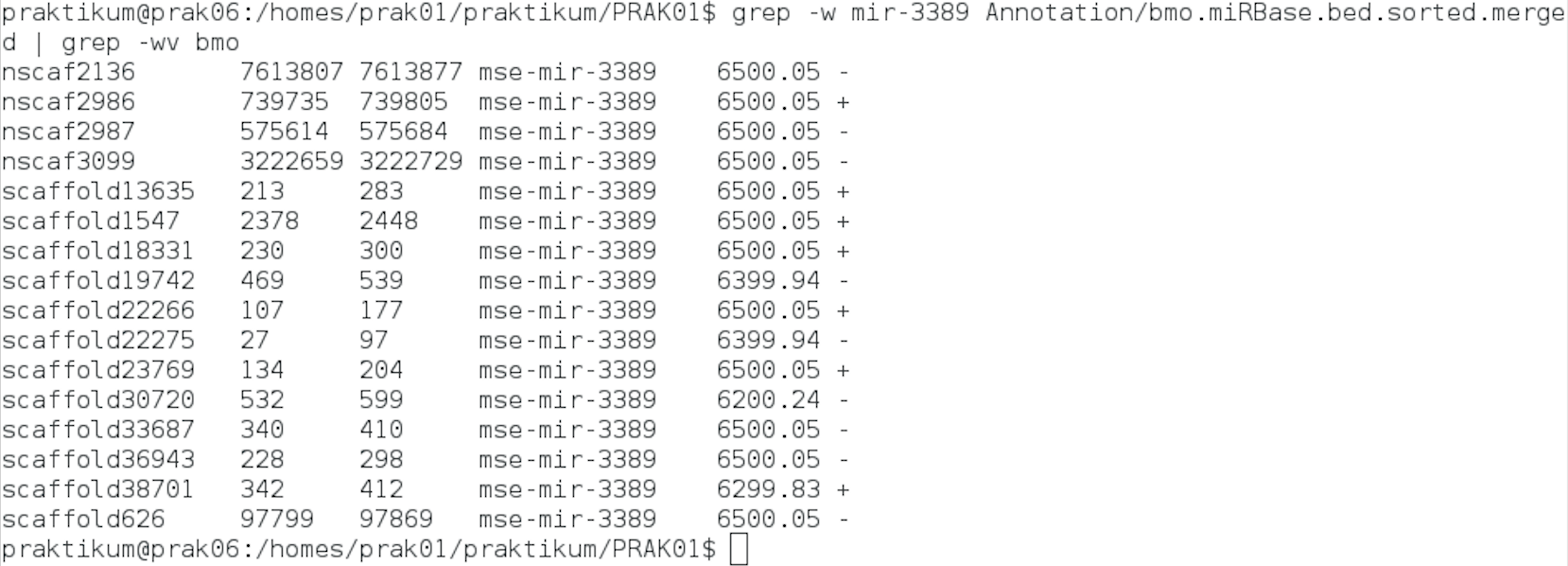
Cluster
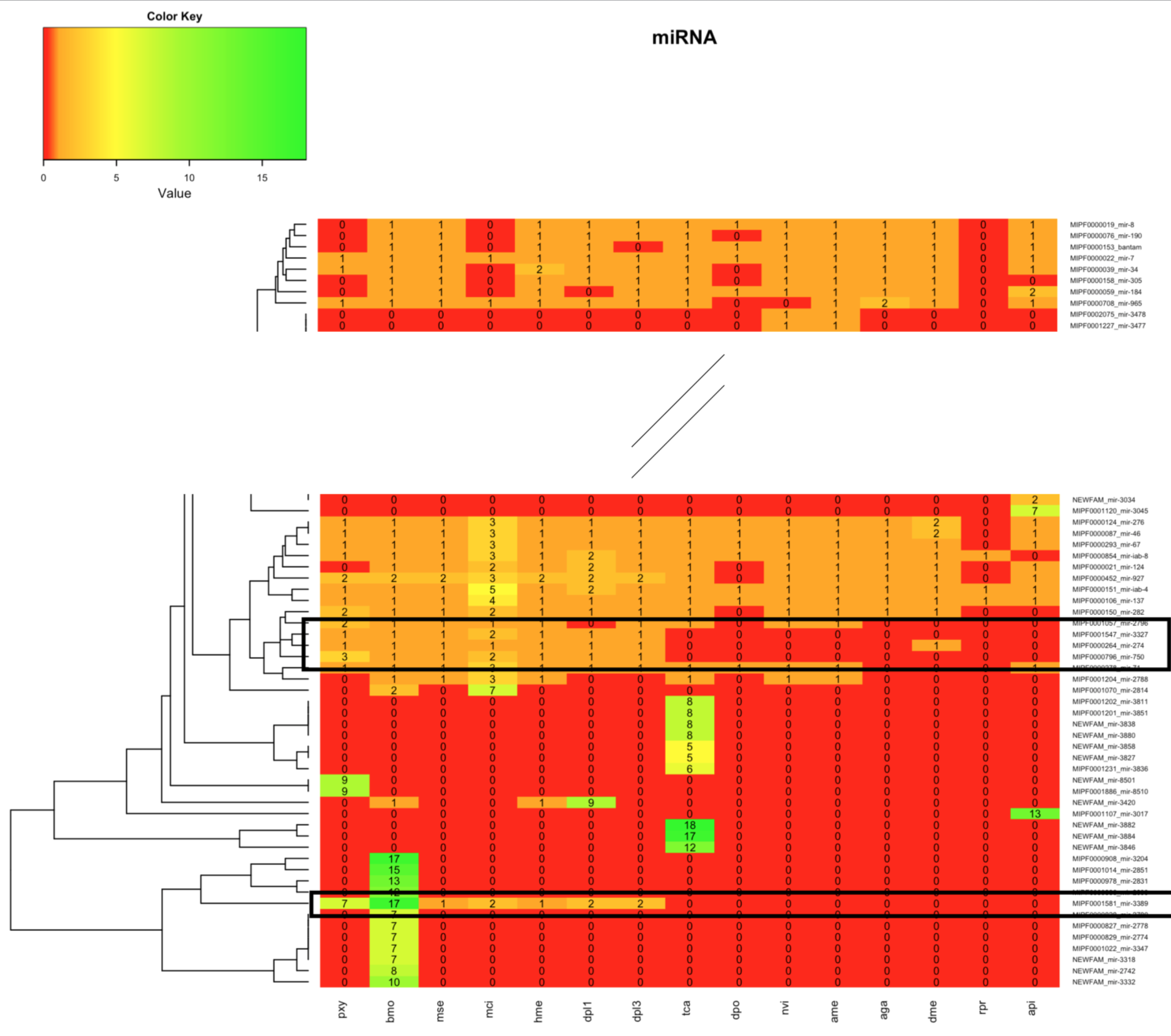
In Heatmap 1.2 waren vor allem die Bereiche im oberen Teil und im unteren Teil des Bildes auffällig. In zwei Bereichen wurden neue miRNAs für einen Großteil der Tiere gefunden. In einem Bereich wurden für alle Lepidoptera neue miRNAs gefunden. Die miRNA-Familien wurden auf der Internetseite von mirbase-Bombix mori auf Cluster überprüft.
Für einzelne miRNAs waren dort Cluster dokumentiert, jedoch keine, die im Dendrogramm benachbart auftauchten.
Datensätze
ursprüngliche Tabelle
Tabelle für Heatmaps
Tabelle für Heatmaps über 30
dpl1.miRBase.bed.sorted.merged
bmo.miRBase.bed.sorted.merged
Skripte
Loss pattern
Die im Verlauf des Praktikums in den untersuchten Spezies gefundenen miRNAs wurden über einen Sequenzvergleich rechnergestützt auf Verwandtschaft untersucht. Aus dem resultierenden File mir_orthodata wurden zunächst aus Gründen der Übersichtlichkeit die miRNAs der sechs Lepidoptera ausgewertet, indem aus dem Vorhandensein der jeweiligen miRNA ein binäres Muster (Pattern) erzeugt wurde, das den Blättern eines phylogenetischen Baumes der sechs Spezies entsprach. Nun konnten die verwandten miRNAs dem jeweiligen Pattern zugeordnet werden. Um die Information von ähnlichen miRNAs mit unterschiedlichen Bezeichnungen (z.B. wurden mir-927 und mir-1926 als Orthologe gefunden) nicht zu verlieren, wurde jeweils ein weiterer Eintrag des anderen Namens erzeugt, der den gleichen Inhalt erhielt, und ebenfalls im jeweiligen Pattern hinterlegt. Die Auswertung und Organisation der Daten erfolgte mithilfe eines Perl-Scriptes.
Die Implementierung der maschinellen Auswertung der sechs Spezies führte zu der Fragestellung, ob dies nicht auch mit gleichwertigem Rechenaufwand für die alle 14 untersuchten Spezies durchführbar wäre. Da ein einfaches Hochskalieren der jeweiligen Prozesse in gleichem Maße auch die Komplexität hinsichtlich Zeit und Speicher in die Höhe getrieben hätte, erforderte das primäre Script wenige Änderungen.
Die für ein sechsstelliges Pattern erforderlichen 64 möglichen Dateien wurden zunächst initial komplett erstellt, die jeweiligen Daten hinzugefügt und schließlich eventuell leere Dateien wieder gelöscht. Allein das Erstellen der Dateien für 14 Spezies (>38000 mögliche Pattern) erforderte ca. 45s. Daher erfolgte eine Umstellung auf die Verwendung von Perl-Subroutines, die das Vorhandensein eines Patterns prüfen und bei Bedarf ein neues File anlegen. Das resultierende Script zeigte im Vergleich zur Vorgängerversion kaum Laufzeiteinbußen.
Die grafische Darstellung des Verlustes von miRNAs während der Artenentwicklung erfolgte über die Erzeugung eines phylogenetischen Baumes. Die Identifizierung der Genverluste konnte durch eine Farbcodierung der Knoten, Äste und Blätter ermöglicht werden.
mir_orthodata Heatmaps














Datensätze
mir_orthodata
miFam.1line.dat
index.pattern kleiner Baum
lossPattern kleiner Baum
index.pattern großer Baum
lossPattern großer Baum
Skripte
patternScript kleiner Baum
patternScript großer Baum
informationScript indexPattern
counterScript Heatmaps
counterScript mir_ortho