
Der Monarchfalter (Danaus plexippus) zählt zu der Ordnung der Lepidoptera und gehört der Familie der Nymphalidae, der Edelfalter, an. Sein Lebensraum liegt vorwiegend in Nordamerika. Es wird in eine westliche und eine östliche Population unterschieden. Die westliche Population lebt zwischen den Rocky Mountains und dem Pazifik. Im Herbst wandert diese Population nach Südwesten an die Küste und im Frühjahr wieder zurück. Im Gegensatz dazu wandert die östliche Population, die zwischen dem Atlantik und den Rocky Mountains lebt, im Herbst vom Norden in den Süden nach Mexiko in die Sierra Nevada. Dort überwintert diese Population und wandert im Frühjahr an die südliche Karibikküste Nordamerikas, wo sie sich dann fortpflanzt und die Parentalgeneration stirbt, wohingegen die Nachwuchsgeneration wieder in den Norden in den alten Lebensraum zurückkehrt. Diese pflanzt sich dort ebenfalls fort, woraufhin die nachfolgende Population im darauffolgenden Herbst wieder an die gleiche Stelle Mexikos zurückkehren wie die Individuen der vorletzten Generation. Es ist zu vermuten, dass diese Wanderroute genetisch abgespeichert ist.
Da der Monarchfalter den Neoptera (Neuflüglern) einzuordnen ist und andere Arten dieser Überordnung ein solches Wanderverhalten nicht aufweisen,
ist es von Interesse, welche Unterschiede auf genetischer Ebene existieren. Um entsprechende Unterschiede zureichend auswerten zu können, ist eine möglichst vollständige Annotation der
Gene in Danaus plexippus notwendig.
Die Aufgabe war es, Gene aufzuspüren, zu denen bisher keine Orthologen in Danaus plexippus annotiert waren.
Um zunächst solche Othologen anderer Insektenarten zu aufzufinden wurde das Tool Proteinortho genutzt.
Das Genom von Danaus plexippus wurde zunächst nach Treffern von Genen verwandter Spezies untersucht, welche keine bekannten Orthologen in Danaus plexippus besitzen.
Entsprechende Gene wurden mittels Proteinortho bestimmt; mit Hilfe des Alignment-Tools BLAT wurde das Sequenzalignment durchgeführt.
Das Danaus-Genom wurde separat von zwei unterschiedlichen Quellen untersucht (MonarchBase und Ensembl).
Es wurden die bekannten Gene folgender Organismen gegen das Genom von Danaus plexippus aligned:
Bombyx mori, Manduca sexta, Melitaea cinxia, Heliconius melpomene,
Tribolium castaneum, Dendroctonus ponderosae, Nasonia vitripennis,
Apis mellifera, Anopheles gambiae, Drosophila melanogaster, Rhodnius prolixus,
Acyrthosiphon pisum und Locusta migratoria.
Der BLAT-Output wurde anschließend gefiltert und geclustert und als Ergebnis nach Abgleich der bestehenden Annotation (GFF-Datei) mögliche neue Annotationen ausgegeben.
Für das Filtern und Clustering sowie den Abgleich mit der GFF-Annotation wurde ein Perl-Skript entwickelt.
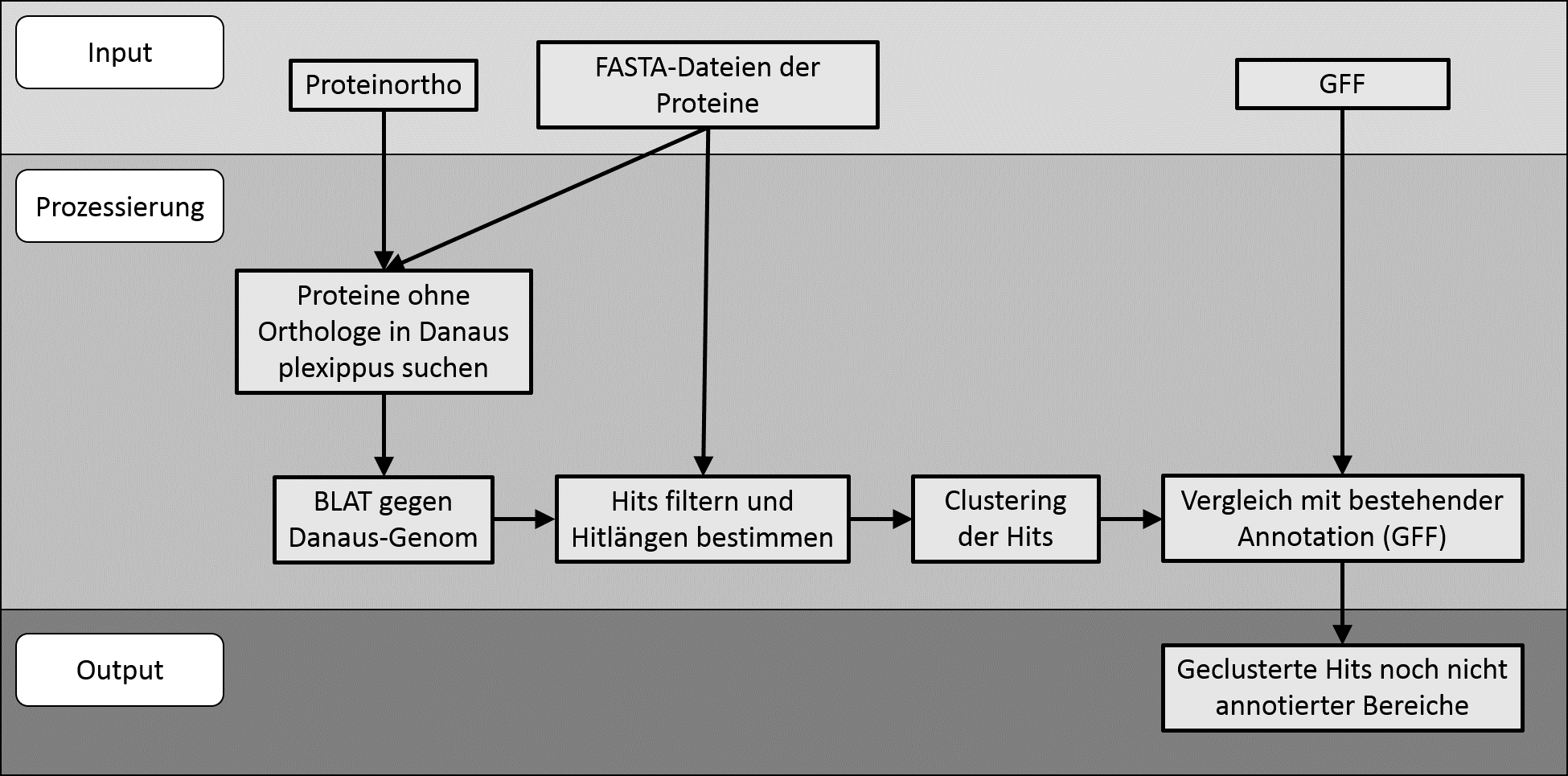
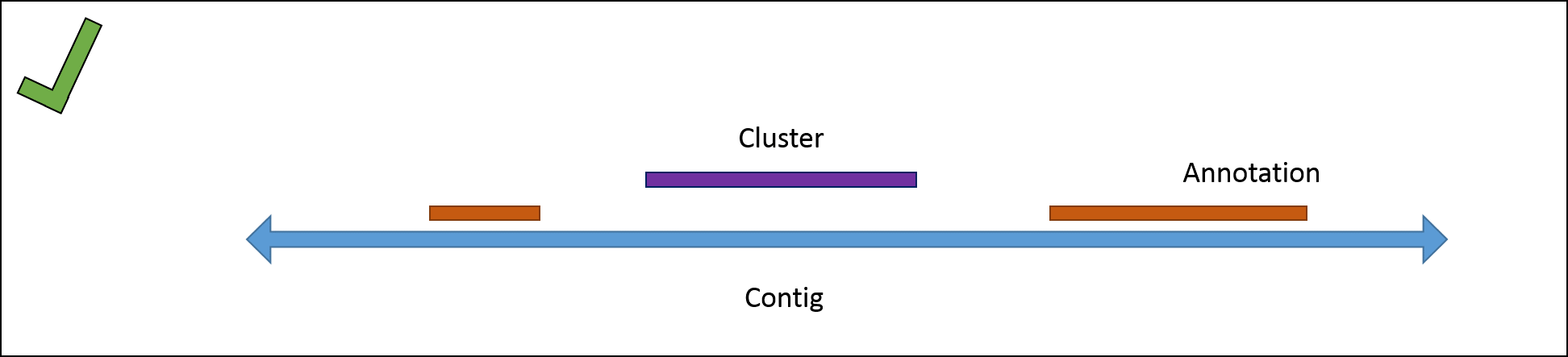
Die Abbildung oben zeigt schematisch die allgemeine Durchführung.
Im Folgenden wird die Durchführung schrittweise besprochen und auf enstprechende Skripte und Output-Dateien verwiesen.
Script: Kompletter Durchlauf
predict_annotation_from_blatout.pl
Mit dem Tool Proteinortho können orthologe Proteine aus verschiedenen Organismen gesucht werden.
Somit wurden Proteine gesucht, zu denen keine bekannten Orthologen in Danaus plexippus existieren.
In folgender Tabelle ist ein schematischer Ausschnitt aus dem Proteinortho-Output gezeigt. Jede Zeile stellt dabei
die von Proteinortho gefundenen Orthologen dar.
| B. mori | D. melanogaster | A. gambiae | D. plexippus (dpl1) | D. plexippus (dpl3) |
|---|---|---|---|---|
| Protein | Protein | * | * | Protein1, Protein2 |
| * | Protein | * | Protein | * |
| Protein | Protein1, Protein2 | Protein | * | * |
| Protein | * | Protein | Protein | Protein |
| * | Protein | Protein | * | * |
| * | * | Protein1, Protein2 | Protein | * |
Es wurden die Zeilen übernommen, in denen keine Orthologen in Danaus plexippus gefunden wurden (grün eingefärbt).
| B. mori | D. melanogaster | A. gambiae | D. plexippus (dpl1) | D. plexippus (dpl3) |
|---|---|---|---|---|
| Protein | Protein1, Protein2 | Protein | * | * |
| * | Protein | Protein | * | * |
Anschließend wurden die Sequenzen aller Proteine dieser Zeilen mit einem Perl-Skript in eine einzige Fasta-Datei übernommen.
Script: Filtern des Proteinortho-Output
filter_proteinortho.pl
Script: Orthologe in eine Fasta-Datei übernehmen
search_fasta.pl
Die Fasta-Datei aus 3.1 wurde mit dem Alignment-Tool BLAT gegen das Genom von Danaus plexippus aligned. Der Output wurde gefiltert, sodass lediglich noch Hits mit einem geringeren e-Value als e-20 ausgegeben wurden. Zusätzlich wurden die Hits mit den Proteinsequenz-Längen und der Strangorientierung versehen. Im Folgenden ist ein Beispiel für einige Zeilen des BLAT-Outputs gegeben:
| Query id | Subject id | %identity | align. length | mismatches | gap openings | q. start | q. end | s. start | s. end | e-value | bit-score | Protein length | Protein %cover | orientation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MCINX001022-PA | DPSCF300208 | 92.92 | 113 | 8 | 0 | 4 | 116 | 55445 | 55107 | 1.3e-59 | 227.0 | 135 | 82 | - |
Script: BLAT-Output filtern
filter_blatout.pl
Script: BLAT-Output erweitern
extend_blatout.pl
Da die BLAT-Hits keine Introns mit einbeziehen und in einigen Fällen die Orthologen nur teilweise Hits im entsprechenden Gen liefern, wurden die
Hits der Orthologen je Contig geclustert, sodass ein Bereich mit einer Start- und Endposition ausgegeben werden kann, in welchem vermutlich ein
Ortholog im Danaus plexippus liegt.
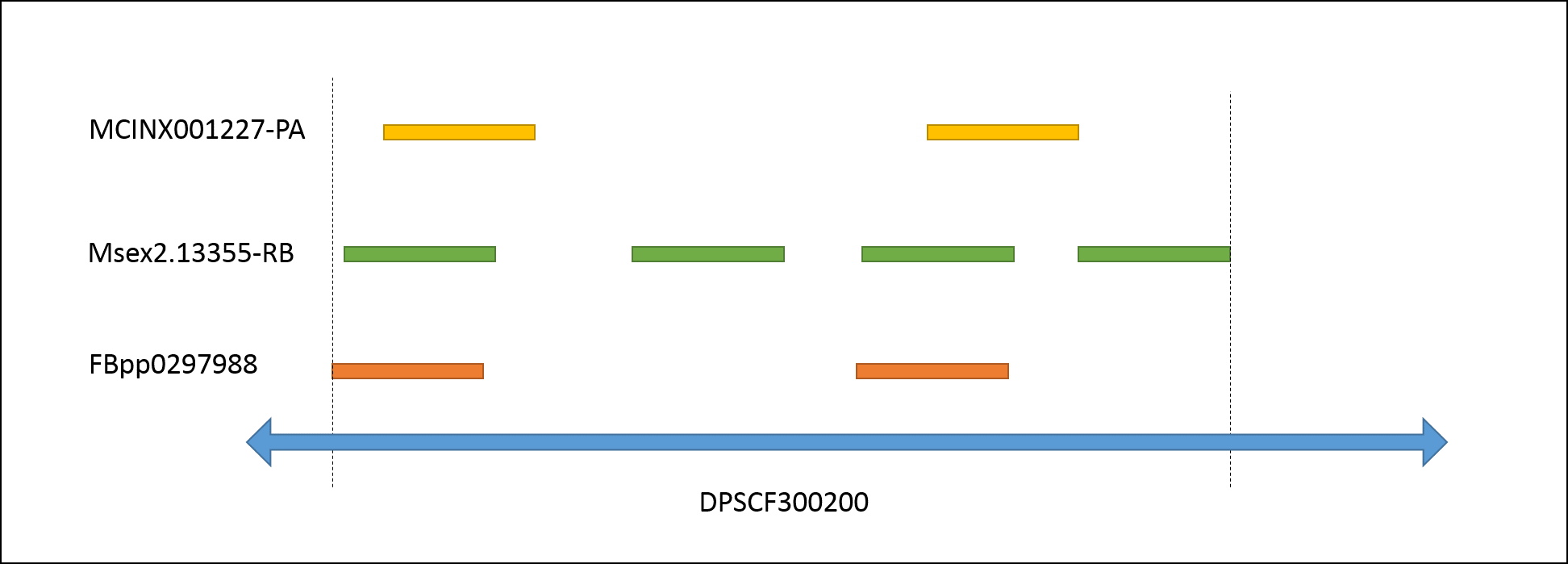
Im Folgenden ist eine BLAT-Output-Datei gezeigt, in welcher mehrere Hits von drei orthologen Proteinen in
einem Contig von Danaus plexippus gefunden wurden. Schematisch sind die Hits in der Abbildung darunter dargestellt.
| Query id | Subject id | %ident. | align. len. | mismatches | gap open. | q. start | q. end | s. start | s. end | e-value | bit-score | CDS length | CDS cover | orient. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MCINX001227-PA | DPSCF300200 | 87.50 | 56 | 7 | 0 | 78 | 133 | 121892 | 122059 | 1.1e-22 | 104.0 | 168 | 32 | + |
| MCINX001227-PA | DPSCF300200 | 87.50 | 56 | 7 | 0 | 78 | 133 | 125533 | 125700 | 1.1e-22 | 104.0 | 168 | 32 | + |
| Msex2.13355-RB | DPSCF300200 | 98.33 | 60 | 1 | 0 | 176 | 235 | 123335 | 123514 | 7.7e-26 | 115.0 | 238 | 24 | + |
| Msex2.13355-RB | DPSCF300200 | 98.33 | 60 | 1 | 0 | 176 | 235 | 126976 | 127155 | 7.7e-26 | 115.0 | 238 | 24 | + |
| Msex2.13355-RB | DPSCF300200 | 82.76 | 58 | 10 | 0 | 72 | 129 | 121892 | 122065 | 1.5e-22 | 104.0 | 238 | 23 | + |
| Msex2.13355-RB | DPSCF300200 | 82.76 | 58 | 10 | 0 | 72 | 129 | 125533 | 125706 | 1.5e-22 | 104.0 | 238 | 23 | + |
| FBpp0297988 | DPSCF300200 | 83.33 | 60 | 10 | 0 | 183 | 242 | 122318 | 122497 | 1.2e-26 | 118.0 | 304 | 19 | + |
| FBpp0297988 | DPSCF300200 | 83.33 | 60 | 10 | 0 | 183 | 242 | 125959 | 126138 | 1.2e-26 | 118.0 | 304 | 19 | + |
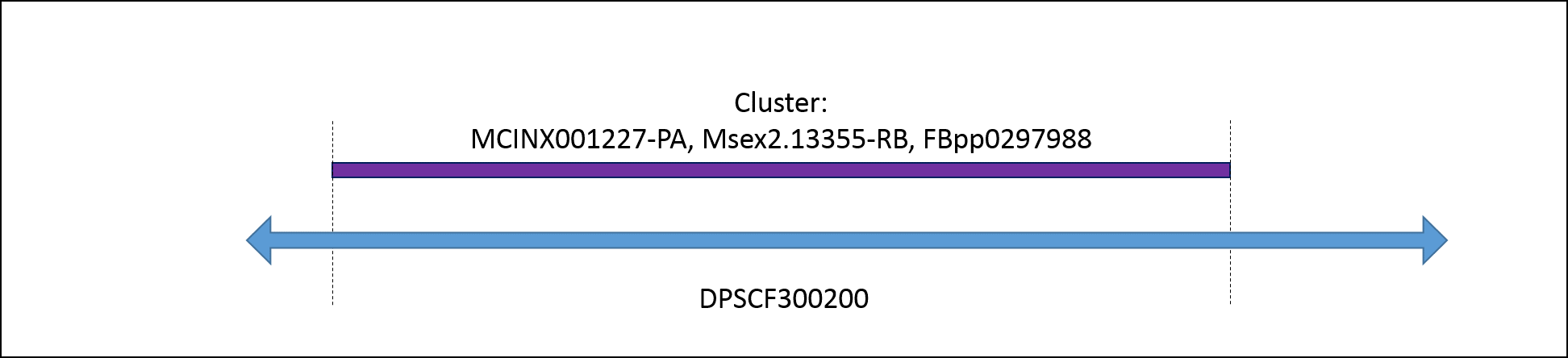
Durch Anwendung eines Perl-Skripts zum Clustering der Hits wird eine Datei mit dem der Untenstehenden Tabelle ausgegeben. Die Schematische Abbildung unter der Tabelle zeigt das Cluster.
| Hits | Contig | Cluster start | Cluster end | Gene-IDs | Cluster Orientation |
|---|---|---|---|---|---|
| 8 | DPSCF300200 | 121892 | 127155 | MCINX001227-PA,Msex2.13355-RB,FBpp0297988 | + |
Script: Clustering der Hits
clustering.pl
Im Anschluss an das Clustering (siehe 3.3) wurden die Clusterpositionen mit den bestehenden Annotationen der GFF-Dateien verglichen.
Dies wurde mit Hilfe eines Perl-Skripts durchgeführt.
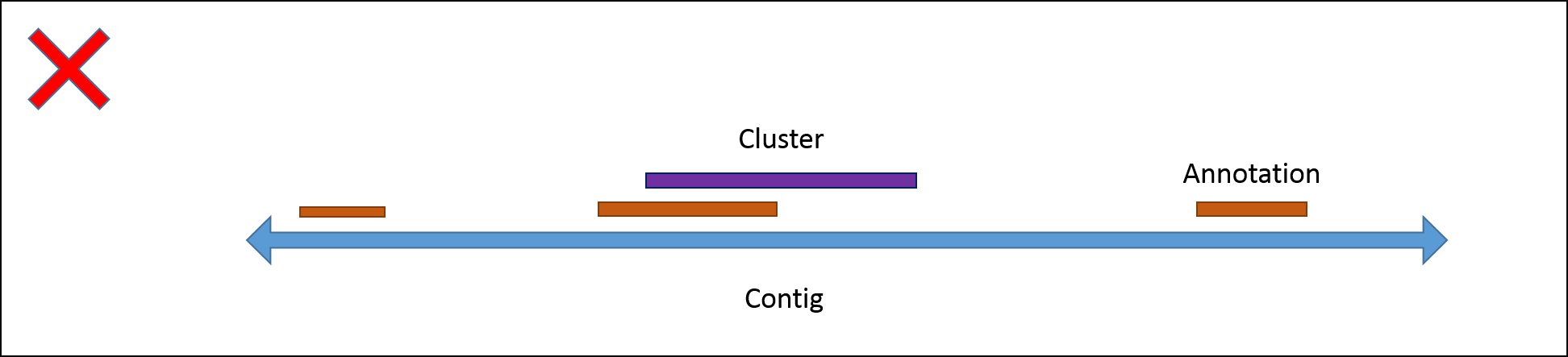
Falls innerhalb des Clusters bereits ein annotierter Bereich existiert, wurde dieser nicht weiter übernommen.
Ein solcher Fall ist im folgenden Bild schematisch dargestellt.
Andernfalls, wenn keine Annotation im Bereich des Clusters gefunden wurde, wird das Cluster übernommen und als ein mögliches neues Ortholog in Danaus plexippus gewertet. Das folgende Bild zeigt einen solchen Fall schematisch:
Der finale Output nach erfolgreichem Vergleich mit der GFF-Annotation ist an einem Beispiel-Cluster in der folgenden Tabelle gezeigt:
| Contig | Cluster start | Cluster end | Cluster orientation | Cluster length | Hits | Gene-IDs |
|---|---|---|---|---|---|---|
| DPSCF300216 | 393488 | 389893 | - | 3595 | 5 | ACYPI000381-PA,CCG001188.1 |
Script: Vergleich mit Annotation
compare_to_annotation.pl
Es sind im Folgenden exemplarisch geclusterte Hits gezeigt, welche an nicht annotierten Bereichen sitzen. Für die Darstellung wurde der Genom-Browser von MonarchBase genutzt.

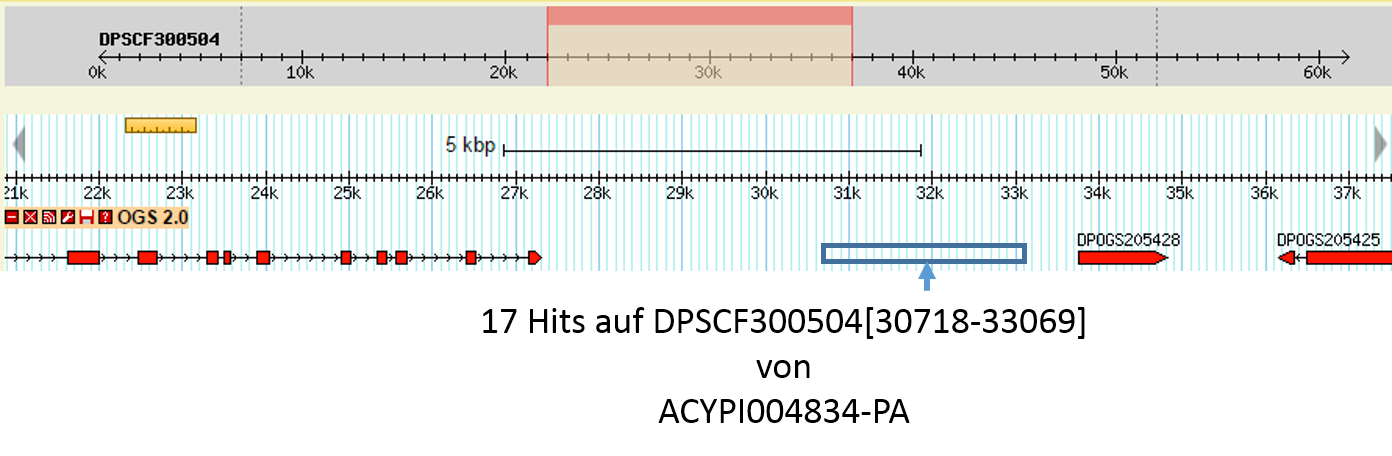
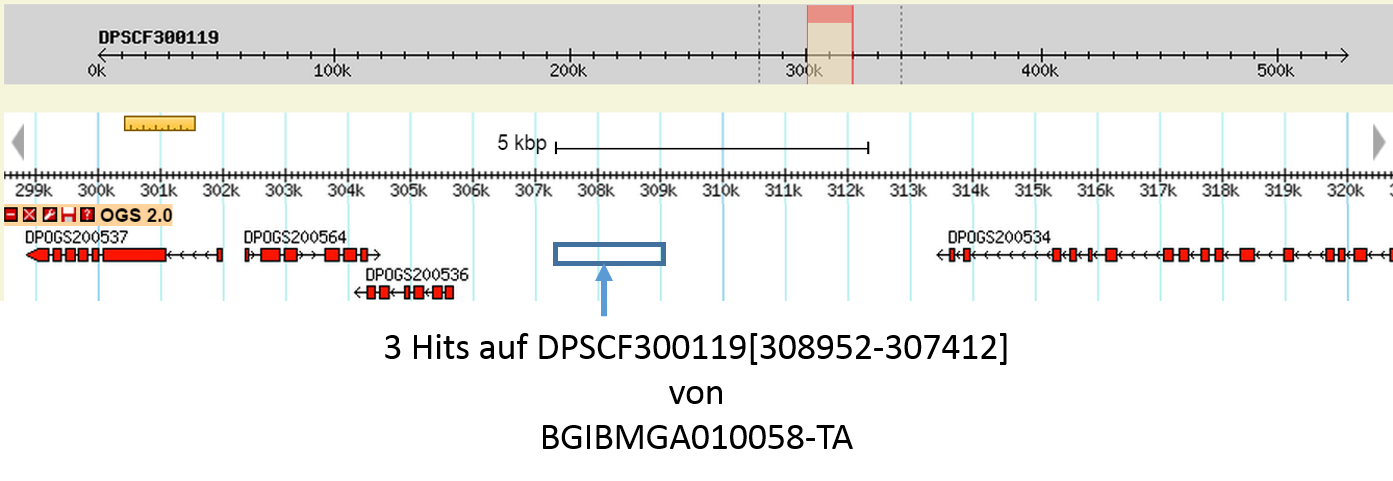
Die Nummern an Hits und unterschiedlichen Orthologen sind für beide Genome (MonarchBase und Ensembl) in den untenstehenden Tabellen gezeigt:
| MonarchBase | ||
|---|---|---|
| Output | Hits | Orthologe |
| Gefiltertes Blatout | 73041 | 8667 |
| Cluster | 12063 | 7200 |
| Nicht annotierte Cluster | 1506 | 258 |
| Ensembl | ||
|---|---|---|
| Output | Hits | Orthologe |
| Gefiltertes Blatout | 80954 | 8671 |
| Cluster | 14958 | 7146 |
| Nicht annotierte Cluster | 904 | 478 |
Ergebnisse:
Gefilterte_Proteinorthotabelle
MonarchBase_Blatout
Ensembl_Blatout
MonarchBase_Extended_Blat_Hits
Ensembl_Extended_Blat_Hits
MonarchBase_Cluster
Ensembl_Cluster
MonarchBase_Orthologe_Vorhersage
Ensembl_Orthologe_Vorhersage
Es konnten erfolgreich Positionen (Cluster) auf dem Genom von Danaus plexippus bestimmt werden, welche auf bisher nicht annotierte Gene
hinweisen. Für das Clustering wurden Vereinfachungen gemacht, wodurch vermutlich einige Hits aussortiert wurden, welche auf weitere mögliche nicht annotierte
Gene in Danaus plexippus hinweisen hätten können.
Eine Verbesserung des Clustering-Algorithmus würde sich für weitere Untersuchungen anbieten.
Im Weiteren ist zu klären, ob es sich bei den gefundenen Positionen tatsächlich um Gene handelt. Möglich wäre ein Vergleich mit
Ab-initio-Vorhersagen. Wenn gesichter ist, dass es sich bei einer Vorhersage um ein Gen handelt, müsste die (funktionelle) Identität geklärt werden.
Die Zuordnung der Proteinfamilien könnte mit Hilfe der Pfam-Datenbank geschehen. Weiterhin wäre ein Vergleich mit homologen Proteinen mit bereits bekannter
Funktion eine Möglichkeit zur Vorhersage der Funktion.
Insbesondere Gene, welche eine Rolle in Orientierungsmechanismen oder der Kontrolle epigenetischer Mechanismen spielen, sind von großem Interesse für die Untersuchung
genetischer Ursachen der Migration.
Weblinks:
http://reppertlab.org/
http://en.wikipedia.org/wiki/Monarch_butterfly
http://metazoa.ensembl.org/index.html
http://monarchbase.umassmed.edu/
Literatur:
Zhan S, Zhang W, Niitepold K, Hsu J, Haeger F, Zalucki MP, Altizer S, de Roode JC, Reppert SM, Kronforst MR (2014).
The genetics of monarch butterfly migration and warning coloration.
Merlin C, Heinze S, Reppert SM (2012).
Unraveling navigational mechanisms in migratory insects.
Zhu H, Gegear RJ, Casselman A, Kanginakudru S, Reppert SM (2009).
Defining behavioral and molecular differences between summer and migratory monarch butterflies.
Zhan S, Reppert SM (2013).
MonarchBase: the monarch butterfly genome database.