Methodik
Phylogenetische Konservierung von IFN-Gamma
Ausgehend von der humanen IFN-Proteinsequenz wurde mittels folgender Proteindatenbanken und Alignment-Tools der Grad der phylogenetischen Konservierung untersucht:
NCBI
UCSC
Ensemble
Mithilfe der dargestellten Software wurden die Informationen der DNA-Datenbanken genutzt, um eine genetische Karte der menschlichen IFN-Gamma-Genstruktur mit der 5' und 3' UTR zu erstellen.
Ermittlung der PKR-Aktivatorsequenz von IFN-Gamma-mRNA aus Spezies der Deuterostomia
Mittels oben genannter Software und Datenbanken wurden ausgehend von der menschlichen Proteinsequenz fuer IFN-Gamma homologe Proteinsequenzen aus fuenf verschiedenen Tierarten (Gepard, Frettchen, Katze, Pavian, Zebra) bestimmt.
Anschliessend wurden diese Proteinsequenzen mit dem Programm tblastn gegen repraesentative Vertreter der Deuterostomia aligniert. Hierbei wurden die Exons des IFN-Gamma-Gens und deren genetische Lokalisation erhalten. Beispiel fuer Panda
Die PKR-Aktivatorsequenz von IFN-Gamma umfasst den kompletten 5'UTR Bereich sowie die ersten 78 Nukleotide des translatierten Bereiches von Exon 1 (Chalamish et al., 2009). Anstelle der kompletten IFN-Gamma mRNA-Sequenz wurde nur dieser Bereich herausgeschnitten und weitergehend untersucht. Mithilfe eines weiteren Programmes, tblastn_pipe, wurden die Ergebnisse gefiltert, indem nur die Ergebnisse identifiziert wurden, die das erste und das letzte Exon mit einem E-value von mindestens 1e-5 beinhalteten.Beste Spezies mit DNA-Bereich
Um den Aktivatorbereich herauszuschneiden, welcher die PKR-Aktivatorsequenz codiert, wurde mittels des Programms get_fastacmd aus den zuvor gefilterten Spezies derjenige DNA-Bereich, welcher 125 Nukleotide stromaufwaerts der Transkriptionsstartstelle des Exon 1 und 78 Nukleotide stromabwaerts liegt, herausgeschnitten. Diese ausgeschnittenen Sequenzen werden als codierende Bereiche der PKR-Aktivatorsequenz angesehen und fuer weitere Strukturanalysen verwendet.
Aktivatorsequenzen
Die ermittelten Aktivatorsequenzen wurden miteinander aligniert. Hierzu wurde das Programm clustal w verwendet. Alignment
Strukturaufklaerung der Aktivatorsequenzen mit bioinformatischen Methoden
Die PKR-Aktivatorsequenz aus verschiedenen Organismen wurde mithilfe verschiedener Programme virtuell gefaltet. Es wurden Programme verwendet, welche einen Vorschlag ueber die Gesamtfaltung des Molekuels lieferten sowie Programme, welche lediglich einzelne Strukturelemente, wie kink turns oder Pseudoknoten vorhersagten.
| Sekundaerstruktur |
Kink-turn |
Pseudoknoten |
| RNAfold |
RMdetect |
pKnotsRG |
| RNAalifold |
|
vsfold5 |
| R-Coffee |
|
|
| locaRNA |
|
|
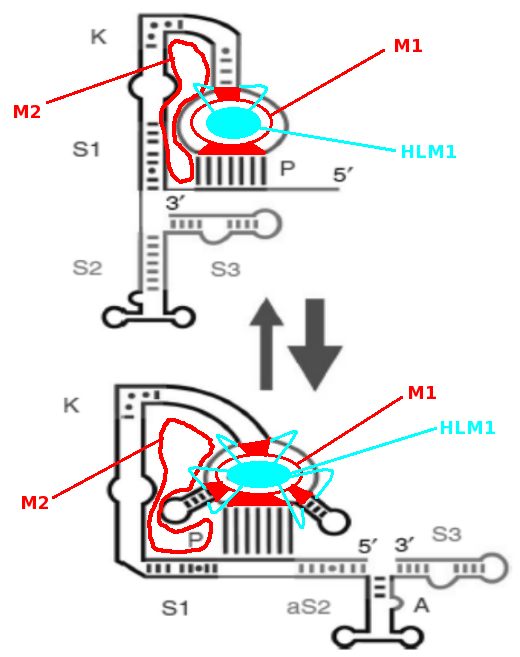
Um die freie Energie der von Chalamish postulierten Aktivatorstruktur mit Pseudoknoten zu berechnen, wurde diese in mehrere Teilstuecke zerlegt, die jeweiligen Energiebeitraege berechnet und aufsummiert. Es wurde folgende Formel verwendet: Sp= S-HLM1+M1+M2+(HL-L)+6
Sp bezeichnet die komplette Struktur mit Pseudoknoten, S die komplette Struktur ohne Pseudoknnoten und die Energie P des Pseudoknoten wurde aus HL-L berechnet. Hl stellt den Pseudoknoten mit Loop dar und L den Loop mit nur einer Basenpaarung. Die anderen Abkuerzungen sind in der Skizze dargestellt. Es wurde ein zusaetzlicher Strafenergiebetrag von 6 kcal/mol dazuaddiert.
{kind=link}