{kind=link}
{kind=link}
Herausschneiden der codierenden Sequenzen durch cut_coding.pl (umbenennen der Dateien 'Banth', 'Bcere', 'Bclau', 'Bholo', 'Blich', 'Bsubt')
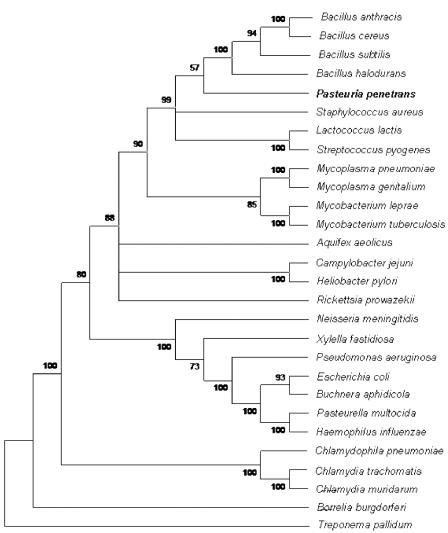
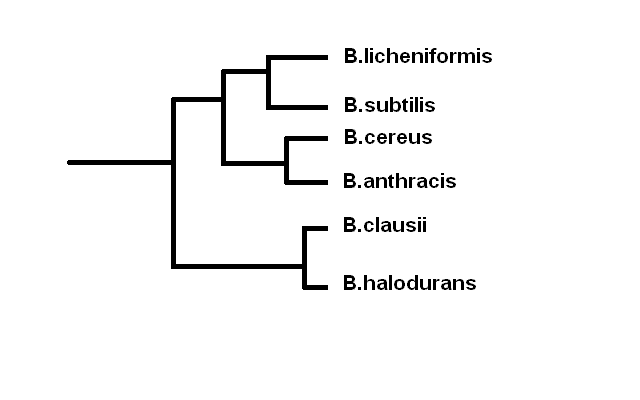
erstellen des phylogenetischen Baumes aus Sekundaerliteratur:
((Bhalo Bclau)((Banth Bcere)(Bsubt Blich)))
Quellen:
erstellen von paarweisen Alignments mittels all_bz durch den phylogenetischen Baum
starten von TBA -> erstellt die Threaded Block Alignments aller Genome (tba.maf)
1. Weg
Erstellen des Scriptes 'mafedit.pl' zum Umwandeln der .maf-Datei in FASTA-Format-Dateien, so dass nur Alignments mit mehr als 20 Basen und wenigstens 3 Sequenzen übernommen werden
Anwenden von 'mafedit.pl' auf die tba.maf-Datei
Anwenden von ClustalW auf die mit 'mafedit.pl' erstellte Datei, um die Alignments zu verbessern
dann trimmen der entstandenen Datei mittels TrimRealignments.pl (Skript von dominic)
Anwendung von Programm RNAZwindow auf getrimmte Datei (Erstellen von Fenstern, (Size = 120, Slide = 40 ) da ab 400 Basen keine Struktur mehr erkennbar)
Anwenden von 'RNAz -b -g' auf die neue Datei um Sekundärstrukturen zu finden
Anwenden von rnazCluster -> erstellen der results.dat
erstellt wurden 150 Loki
RNAzBLAST gestartet mit /NONCODE und mit /Rfam mit den results von RNAzCLUSTER
2. Weg
Anwendung von Programm RNAZwindow auf die tba.maf Datei (Erstellen von Fenstern, (Size = 120, Slide = 40 ) da ab 400 Basen keine Struktur mehr erkennbar)
Anwenden von 'RNAz -b -g' auf das Ergebnis
Anwenden von rnazCluster -> erstellen der results.dat
erstellt 64 Loki
RNAzBLAST gestartet mit /NONCODE und dann mit /Rfam mit den results von RNAzCLUSTER, im Anschluss wurde das Ergebnis mit den vorhandenen *.frn Files aus der NCBI Datenbank geblastet, um schon bekannte RNA Sequenzen zu annotieren.
TRNAscan
Anwenden von tRNAscan auf die von TBA erstellte datei und den OriginalOutput des all_bz
Original: 500 RNA-Schnipsel
TBA: 304 RNA-Schnipsel
ABER: 174 gleiche RNA-Schnipsel