

Next: Verwendete Tools
Up: Vorgehensweise
Previous: Prinzipelle Vorgehensweise
Folgende Schritte haben wir in der Regel abgearbeitet:
- Es wird das Alignment der gegebene Aminosäurestruktur gegen andere Strukturen aus einer Proteindatenbank gebildet. Das Alignment gibt dann Auskunft darüber, ob in dieser Datenbank bereits eine ähnliche, homologe Aminosäuresequenz gespeichert ist.
[blastp; http://www.ncbi.nlm.nih.gov/BLAST/ ; National Center for Biotechnology Information
- Ist ein guter Score zwischen der zu untersuchenden Aminosäuresequenz und einer Sequenz aus der Datenbank ereicht wurden, kann man davon ausgehen, dass es sich dabei um homologe Proteine handelt. Anhand einer weiteren Datenbank ist es nun möglich für das soeben gefundene homologe Protein die Tertiärstruktur als .pdb-File, herunterzuladen.
Protein Data Bank: http://www.rcsb.org/pdb/; Research Collaboratory for Structural Bioinformatics (RCSB)
- Wurde kein Alignment mit einem guten Alignment-Score ermittelt, so wird versucht mittels Fold Recognation eine passende Struktur zu finden. Die Aminosäuresequenz wird hierbei gegen bereits bekannte 3D-Strukturen in einer Datenbank geprüft, bis ein Protein mit einer zu unserer Sequenz passenden Struktur gefunden wird. Das Ergebnis ist dann wieder ein homologes Protein, dessen Tertiärstruktur man aus der Protein Data Bank erhŠlt.
Tool: FUGUE - http://www-cryst.bioc.cam.ac.uk/fugue/
Tool: 3dpssm;
- Wurde weder durch Schritt 1, noch durch die Schritt 3 ein homologes Protein gefunden, so bleibt nur die Auswertung der SekundŠrstrukturen.
- Die .pdb-Datei wird genutzt, um ein Alignment zwischen der zu analysierenden Aminosäuresequenz und der Struktur des homologen Proteins zu erzeugen. Dazu verwenden wir das Internet Tool FUGUE. Als Ergebnis erhält man ein .pir-File für den nächsten Schritt.
Tool: FUGUE - http://www-cryst.bioc.cam.ac.uk/fugue/ ; University of Cambridge, Department of Biochemistry
- Als nächstes erstellen wir mittels MODELLER ein 3D Modell der Tertiärsruktur für die zu untersuchende Aminosäuresequenz. Zur Berechnung benötigt das Programm drei Dateien. Neben der Datei model.top , die Konfigurationszwecken dient sind dies noch zwei Strukturdaten enthaltende Dateien. Zum einen wird das in Schritt 2 erstellte .pir-File benötigt und zum anderen das .pdb-File mit der Tertiärstruktur des homologen Proteins. Anhand dieses Strukturmodells wird nun für die zu analysierende Sequenz die Tertiärstruktur gebildet. Man erhält die Tertiärstruktur in einem .pdb-File.
Tool: Modeller; http://structbio.vanderbilt.edu/comp/soft/modeller/; Vanderbilt University; Ceter fr Structural Biology
- Um die Aussagekraft der gewonnenen Tertiärstruktur zu prüfen wird das erstellte Modell nun noch zwei Prüfungen unterzogen. Das Programm PROFILER berechnet aus den beiden Tertiärstrukturfiles, dem Erstellten und dem Homologen, ihren RMS-Wert (mittlerer quadratischer Fehler). Dieser gibt dann nach folgendem Schlźssel Auskunft über die Korrektheit des erstellten 3D-Modells:
- RMS-Wert
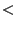 10 3D-Modell ist brauchbar
10 3D-Modell ist brauchbar
- RMS-Wert
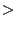 10 3D-Modell ist unbrauchbar
10 3D-Modell ist unbrauchbar
In unserer Auswertung ziehen wir diese Grenze jedoch nicht so eng, sondern betrachten RMS-Werte von bis zu 14 als gut.
Tool: Profiler
- Ein weiteres Tool berechnet nun noch Energiekurven für das erstellte und das homologe Model. Daraus wird ersichtilich, wie stabil das erstellte Modell ist. Des geschieht anhand sogenannter knowledge-based potentials.
Tool: Prosa II - http://lore.came.sbg.ac.at:8080/CAME/CAMEEXTERN/PROSA ; Center of Applied Molecular Engineering, Institute of Chemestry and Biochemistry, University of Salzburg
Next: Verwendete Tools
Up: Vorgehensweise
Previous: Prinzipelle Vorgehensweise
axel
2004-10-29